

Divergent Processing of Cell Stress Signals as the Basis of Cancer Progression: Licensing NFκB on Chromatin
- PMID: 39201306
- PMCID: PMC11354898
- DOI: 10.3390/ijms25168621
Divergent Processing of Cell Stress Signals as the Basis of Cancer Progression: Licensing NFκB on Chromatin
Abstract
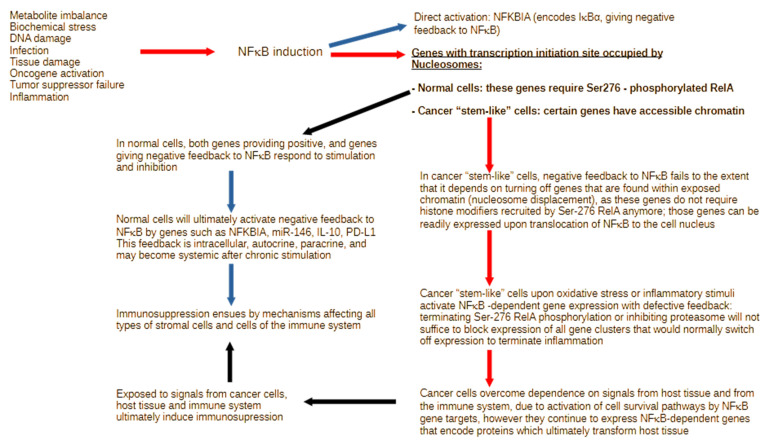
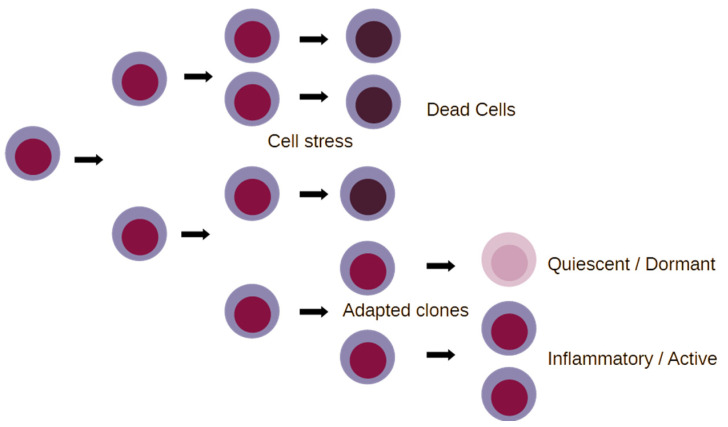
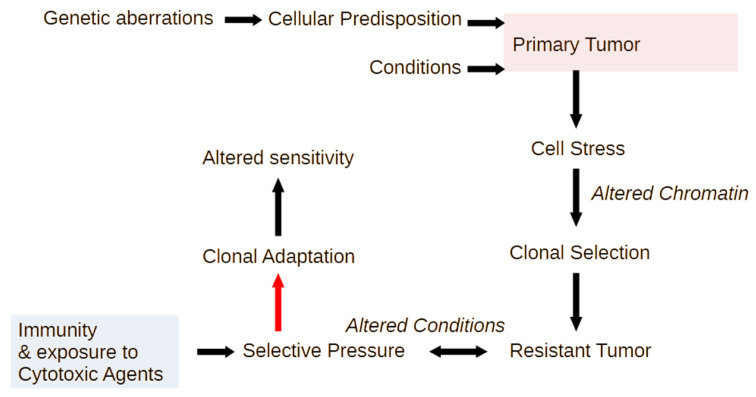
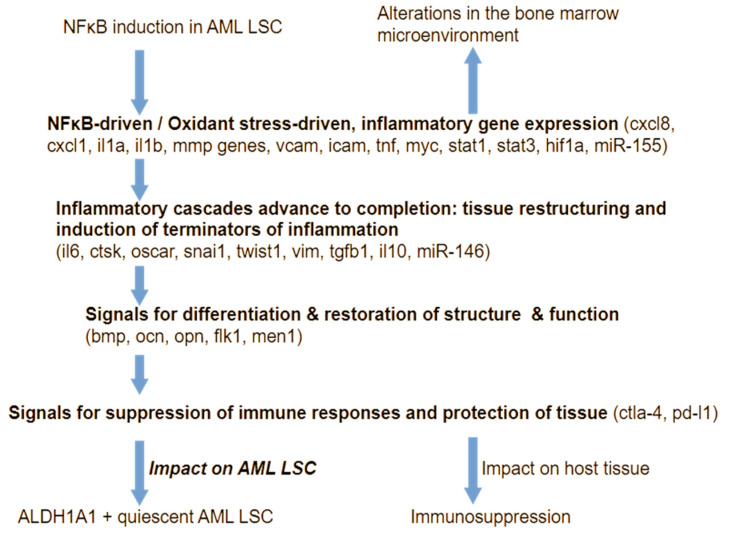
Inflammation is activated by diverse triggers that induce the expression of cytokines and adhesion molecules, which permit a succession of molecules and cells to deliver stimuli and functions that help the immune system clear the primary cause of tissue damage, whether this is an infection, a tumor, or a trauma. During inflammation, short-term changes in the expression and secretion of strong mediators of inflammation occur, while long-term changes occur to specific groups of cells. Long-term changes include cellular transdifferentiation for some types of cells that need to regenerate damaged tissue, as well as death for specific immune cells that can be detrimental to tissue integrity if they remain active beyond the boundaries of essential function. The transcriptional regulator NFκB enables some of the fundamental gene expression changes during inflammation, as well as during tissue development. During recurrence of malignant disease, cell stress-induced alterations enable the growth of cancer cell clones that are substantially resistant to therapeutic intervention and to the immune system. A number of those alterations occur due to significant defects in feedback signal cascades that control the activity of NFκB. Specifically, cell stress contributes to feedback defects as it overrides modules that otherwise control inflammation to protect host tissue. NFκB is involved in both the suppression and promotion of cancer, and the key distinctive feature that determines its net effect remains unclear. This paper aims to provide a clear answer to at least one aspect of this question, namely the mechanism that enables a divergent response of cancer cells to critical inflammatory stimuli and to cell stress in general.
Keywords: BET inhibitor; cell stress; chemokines; chromatin; cytokines; histone; inflammation; nuclear factor kappa B; super enhancer; unfolded protein response.
Conflict of interest statement
The author declares no conflict of interest.
Figures
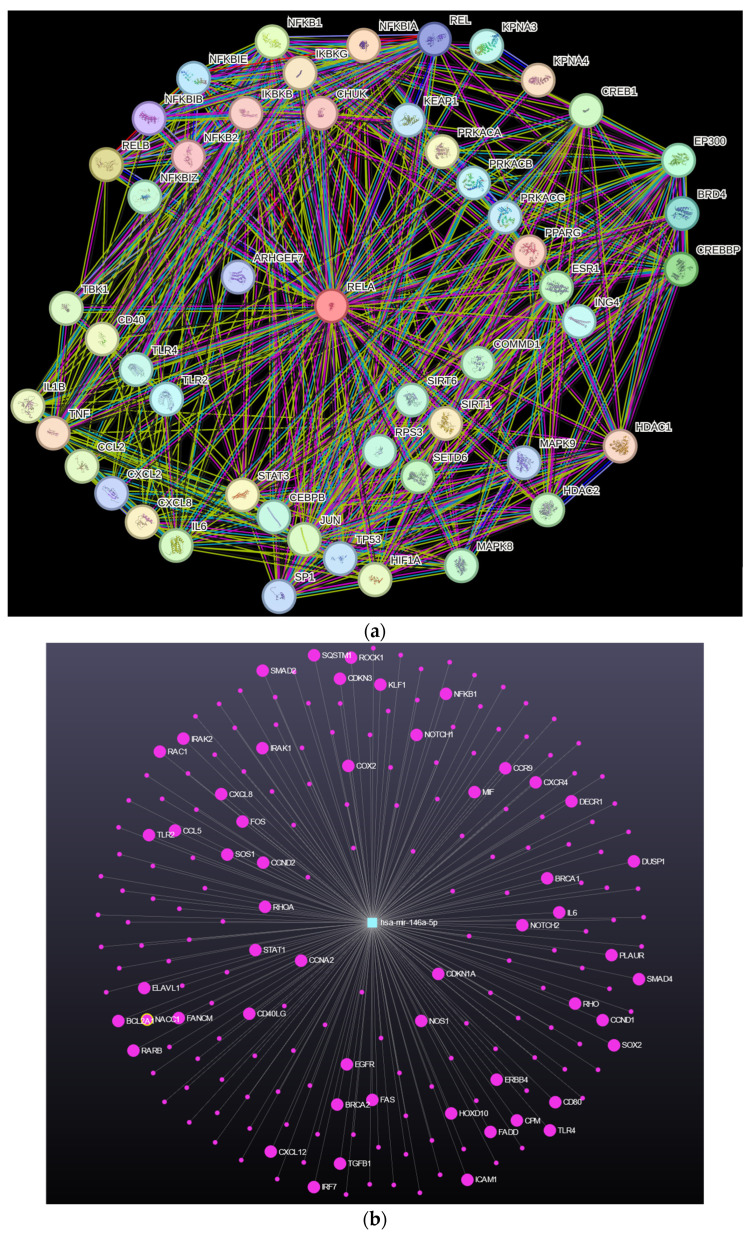
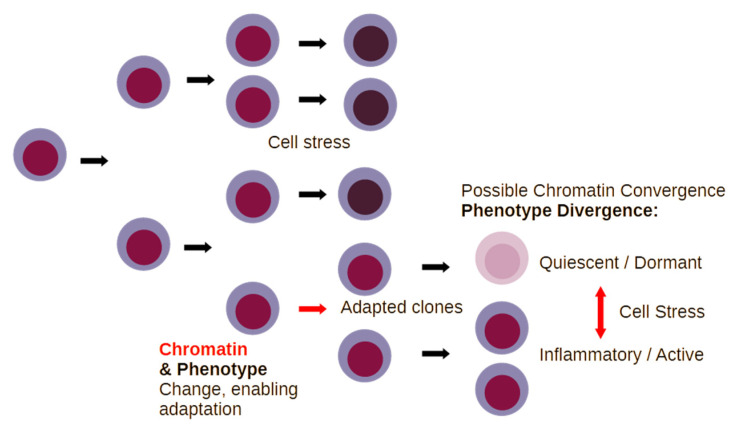
References
-
- Lu H., Chen R., Barnie P.A., Tian Y., Zhang S., Xu H., Chakrabarti S., Su Z. Fibroblast Transdifferentiation Promotes Conversion of M1 Macrophages and Replenishment of Cardiac Resident Macrophages Following Cardiac Injury in Mice. Eur. J. Immunol. 2020;50:795–808. doi: 10.1002/eji.201948414. - DOI - PubMed
-
- Naitoh H., Suganuma Y., Ueda Y., Sato T., Hiramuki Y., Fujisawa-Sehara A., Taketani S., Araki M. Upregulation of Matrix Metalloproteinase Triggers Transdifferentiation of Retinal Pigmented Epithelial Cells in Xenopus Laevis: A Link between Inflammatory Response and Regeneration. Dev. Neurobiol. 2017;77:1086–1100. doi: 10.1002/dneu.22497. - DOI - PubMed
-
- Gaba A., Grivennikov S.I., Do M.V., Stumpo D.J., Blackshear P.J., Karin M. Cutting Edge: IL-10-Mediated Tristetraprolin Induction Is Part of a Feedback Loop That Controls Macrophage STAT3 Activation and Cytokine Production. J. Immunol. 2012;189:2089–2093. doi: 10.4049/jimmunol.1201126. - DOI - PMC - PubMed
-
- Stedile M., Lara Montero A., García Solá M.E., Goddio M.V., Beckerman I., Bogni E., Ayre M., Naguila Z., Coso O.A., Kordon E.C. Tristetraprolin Promotes Survival of Mammary Progenitor Cells by Restraining TNFα Levels. Front. Cell Dev. Biol. 2023;11:1265475. doi: 10.3389/fcell.2023.1265475. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
