

Ubiquitination Insight from Spinal Muscular Atrophy-From Pathogenesis to Therapy: A Muscle Perspective
- PMID: 39201486
- PMCID: PMC11354275
- DOI: 10.3390/ijms25168800
Ubiquitination Insight from Spinal Muscular Atrophy-From Pathogenesis to Therapy: A Muscle Perspective
Abstract
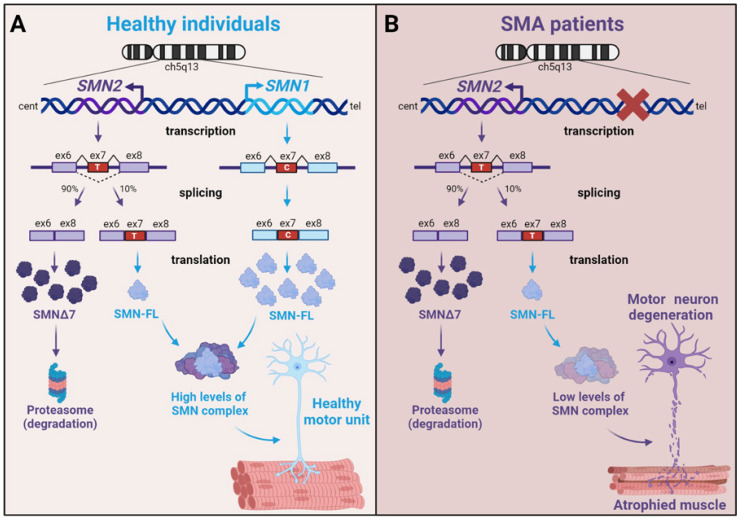
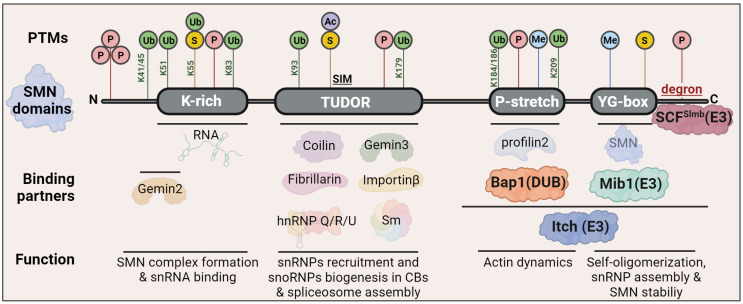
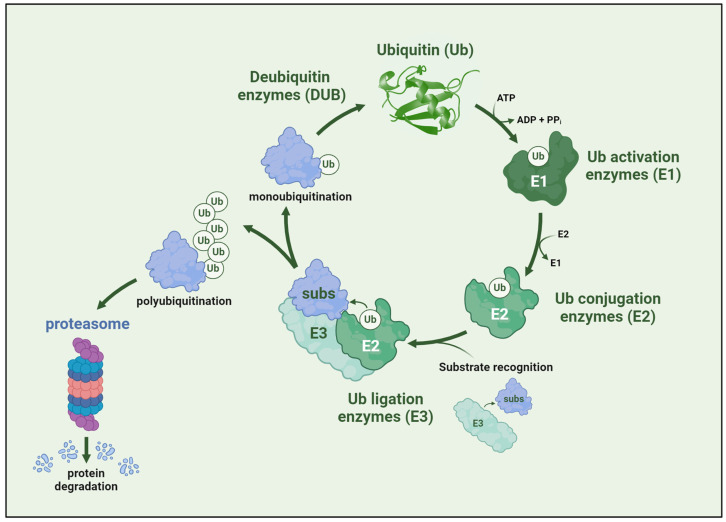
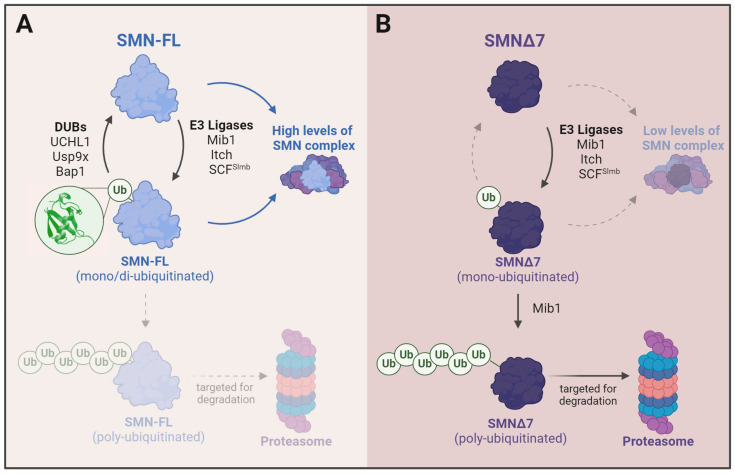
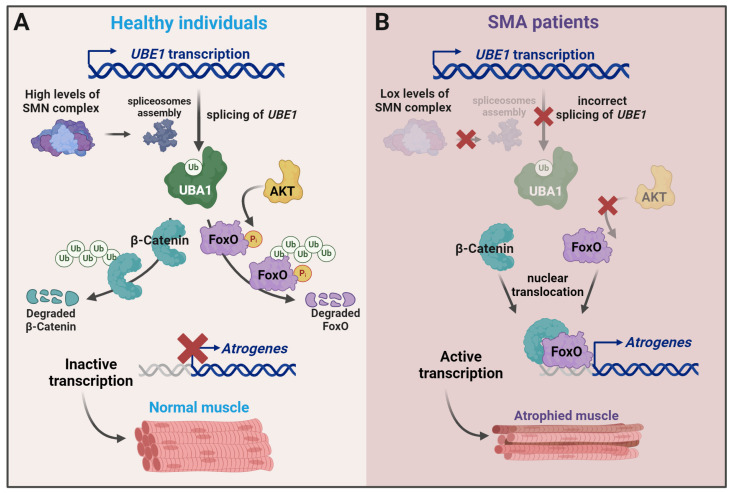
Spinal muscular atrophy (SMA) is one of the most frequent causes of death in childhood. The disease's molecular basis is deletion or mutations in the SMN1 gene, which produces reduced survival motor neuron protein (SMN) levels. As a result, there is spinal motor neuron degeneration and a large increase in muscle atrophy, in which the ubiquitin-proteasome system (UPS) plays a significant role. In humans, a paralogue of SMN1, SMN2 encodes the truncated protein SMNΔ7. Structural differences between SMN and SMNΔ7 affect the interaction of the proteins with UPS and decrease the stability of the truncated protein. SMN loss affects the general ubiquitination process by lowering the levels of UBA1, one of the main enzymes in the ubiquitination process. We discuss how SMN loss affects both SMN stability and the general ubiquitination process, and how the proteins involved in ubiquitination could be used as future targets for SMA treatment.
Keywords: SMN; skeletal muscle atrophy; spinal muscular atrophy; ubiquitin–proteasome system.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Brzustowicz L.M., Lehner T., Castilla L.H., Penchaszadeh G.K., Wilhelmsen K.C., Daniels R., Davies K.E., Leppert M., Ziter F., Wood D., et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature. 1990;344:540–541. doi: 10.1038/344540a0. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
