

Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets
- PMID: 39201721
- PMCID: PMC11354906
- DOI: 10.3390/ijms25169035
Podocyte Death in Diabetic Kidney Disease: Potential Molecular Mechanisms and Therapeutic Targets
Abstract
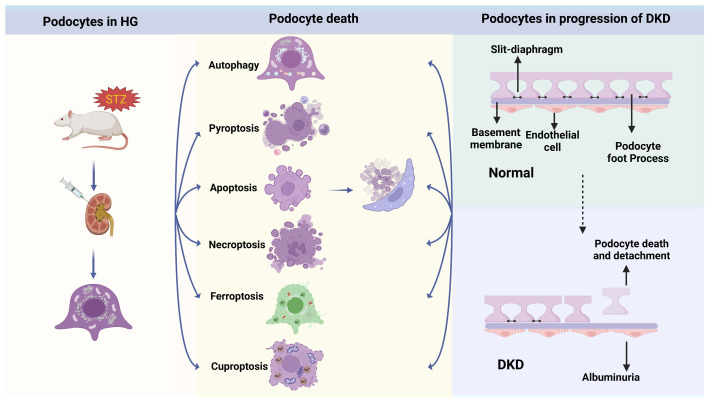
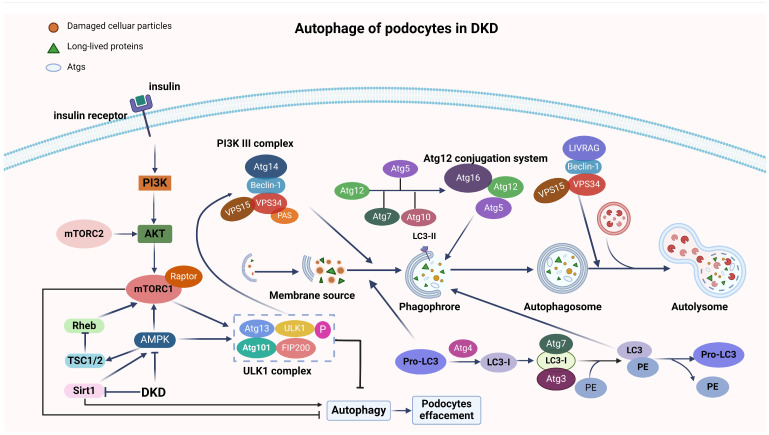
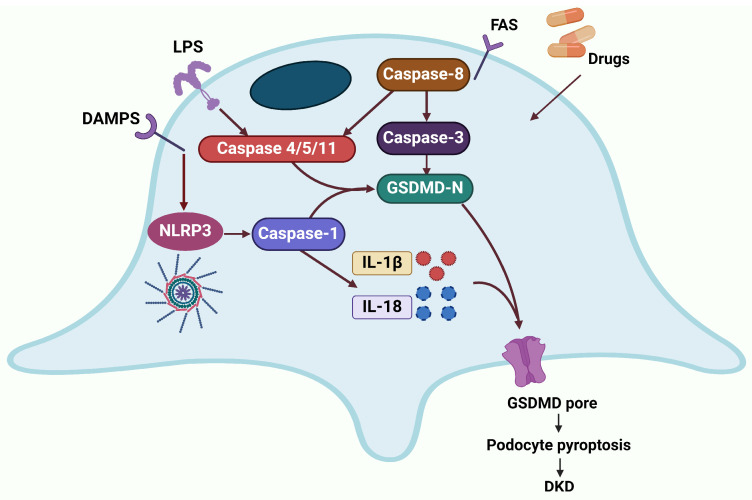
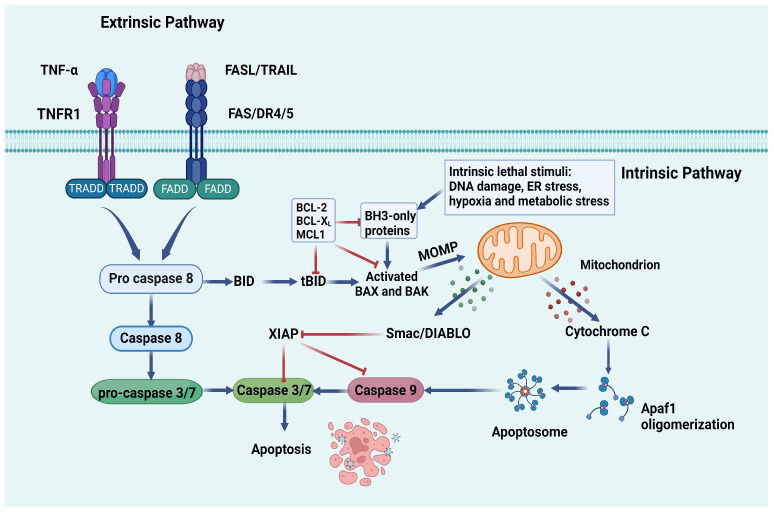
Cell deaths maintain the normal function of tissues and organs. In pathological conditions, the abnormal activation or disruption of cell death often leads to pathophysiological effects. Diabetic kidney disease (DKD), a significant microvascular complication of diabetes, is linked to high mortality and morbidity rates, imposing a substantial burden on global healthcare systems and economies. Loss and detachment of podocytes are key pathological changes in the progression of DKD. This review explores the potential mechanisms of apoptosis, necrosis, autophagy, pyroptosis, ferroptosis, cuproptosis, and podoptosis in podocytes, focusing on how different cell death modes contribute to the progression of DKD. It recognizes the limitations of current research and presents the latest basic and clinical research studies targeting podocyte death pathways in DKD. Lastly, it focuses on the future of targeting podocyte cell death to treat DKD, with the intention of inspiring further research and the development of therapeutic strategies.
Keywords: apoptosis; autophagy; cell death; diabetic kidney disease; ferroptosis; necroptosis; podocyte; pyroptosis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Podocyte programmed cell death in diabetic kidney disease: Molecular mechanisms and therapeutic prospects.Biomed Pharmacother. 2024 Aug;177:117140. doi: 10.1016/j.biopha.2024.117140. Epub 2024 Jul 16. Biomed Pharmacother. 2024. PMID: 39018872 Review.
-
Research progress on multiple cell death pathways of podocytes in diabetic kidney disease.Mol Med. 2023 Oct 12;29(1):135. doi: 10.1186/s10020-023-00732-4. Mol Med. 2023. PMID: 37828444 Free PMC article. Review.
-
Modes of podocyte death in diabetic kidney disease: an update.J Nephrol. 2022 Jul;35(6):1571-1584. doi: 10.1007/s40620-022-01269-1. Epub 2022 Feb 24. J Nephrol. 2022. PMID: 35201595 Review.
-
TRAIL induces podocyte PANoptosis via death receptor 5 in diabetic kidney disease.Kidney Int. 2025 Feb;107(2):317-331. doi: 10.1016/j.kint.2024.10.026. Epub 2024 Nov 19. Kidney Int. 2025. PMID: 39571905
-
JAK/STAT pathway promotes the progression of diabetic kidney disease via autophagy in podocytes.Eur J Pharmacol. 2021 Jul 5;902:174121. doi: 10.1016/j.ejphar.2021.174121. Epub 2021 Apr 24. Eur J Pharmacol. 2021. PMID: 33901462
Cited by
-
The emerging role of cuproptosis in spinal cord injury.Front Immunol. 2025 Jun 16;16:1595852. doi: 10.3389/fimmu.2025.1595852. eCollection 2025. Front Immunol. 2025. PMID: 40589743 Free PMC article. Review.
-
HDAC4: an emerging target in diabetes mellitus and diabetic complications.Eur J Med Res. 2025 May 30;30(1):429. doi: 10.1186/s40001-025-02697-y. Eur J Med Res. 2025. PMID: 40448151 Free PMC article. Review.
-
Natural products in treating diabetic kidney disease: a visualized bibliometric analysis.Front Pharmacol. 2025 May 19;16:1522074. doi: 10.3389/fphar.2025.1522074. eCollection 2025. Front Pharmacol. 2025. PMID: 40458808 Free PMC article.
-
LCZ696 improves oxidative stress injury in human podocytes induced by increased glucose levels via Nrf2/HO-1 signaling pathway.Eur J Med Res. 2025 Jul 9;30(1):598. doi: 10.1186/s40001-025-02883-y. Eur J Med Res. 2025. PMID: 40635044 Free PMC article.
References
-
- Tuttle K.R., Bakris G.L., Bilous R.W., Chiang J.L., De Boer I.H., Goldstein-Fuchs J., Hirsch I.B., Kalantar-Zadeh K., Narva A.S., Navaneethan S.D., et al. Diabetic Kidney Disease: A Report froman ADA Consensus Conference. Diabetes Care. 2014;37:2864–2883. doi: 10.2337/dc14-1296. - DOI - PMC - PubMed
-
- Bikbov B., Purcell C.A., Levey A.S., Smith M., Abdoli A., Abebe M., Adebayo O.M., Afarideh M., Agarwal S.K., Agudelo-Botero M., et al. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395:709–733. doi: 10.1016/S0140-6736(20)30045-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
