

Comprehensive Analysis of Methylome and Transcriptome to Identify Potential Genes Regulating Porcine Testis Development
- PMID: 39201790
- PMCID: PMC11354776
- DOI: 10.3390/ijms25169105
Comprehensive Analysis of Methylome and Transcriptome to Identify Potential Genes Regulating Porcine Testis Development
Abstract
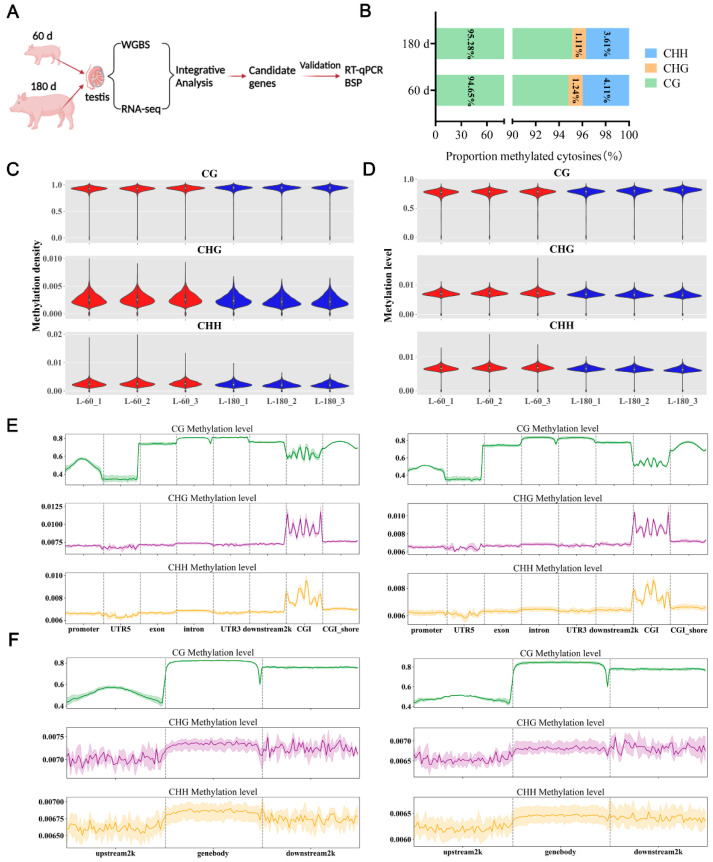
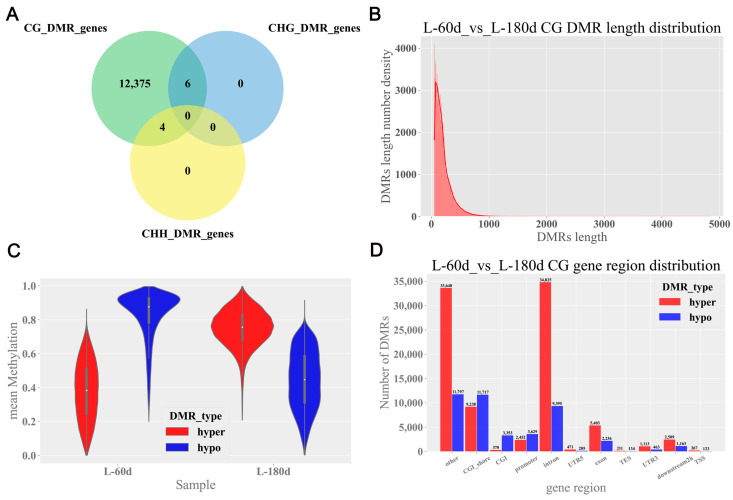
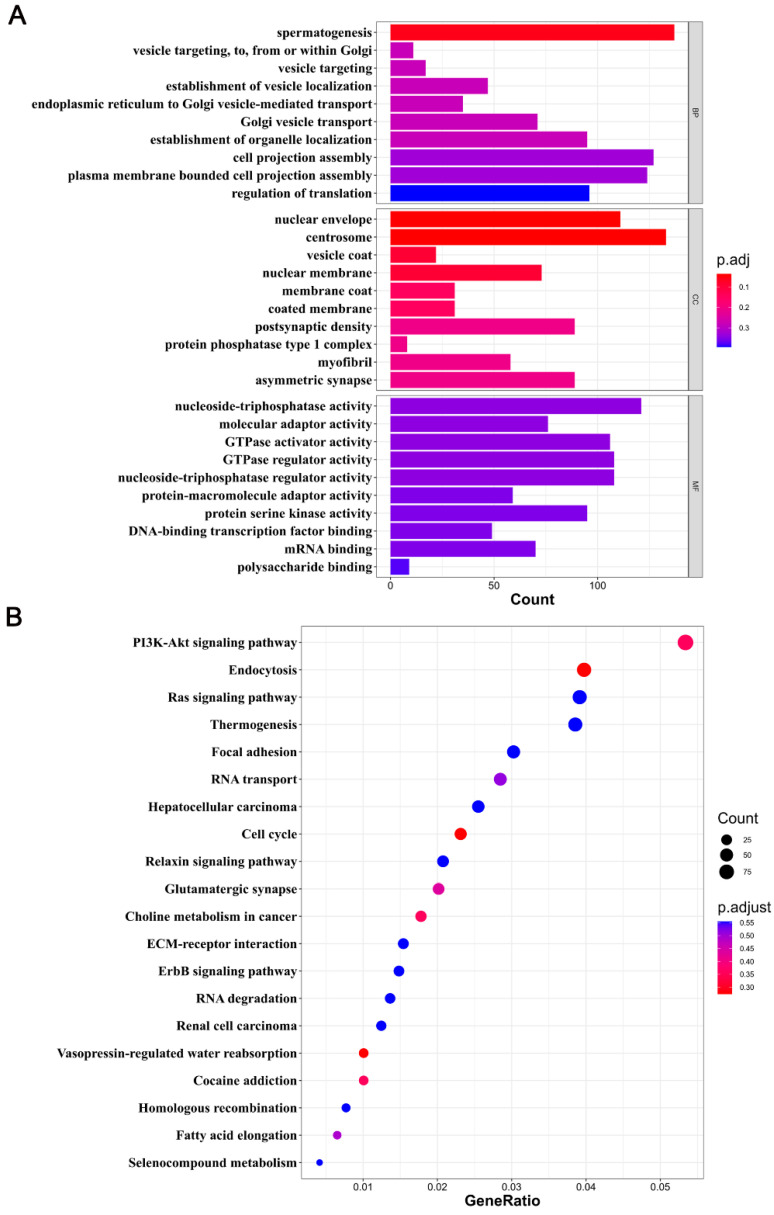
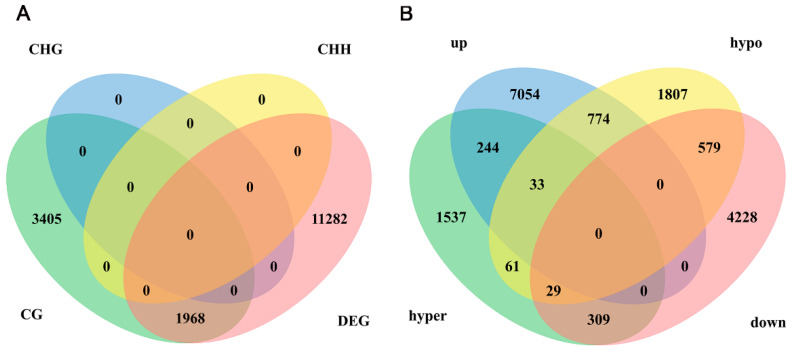
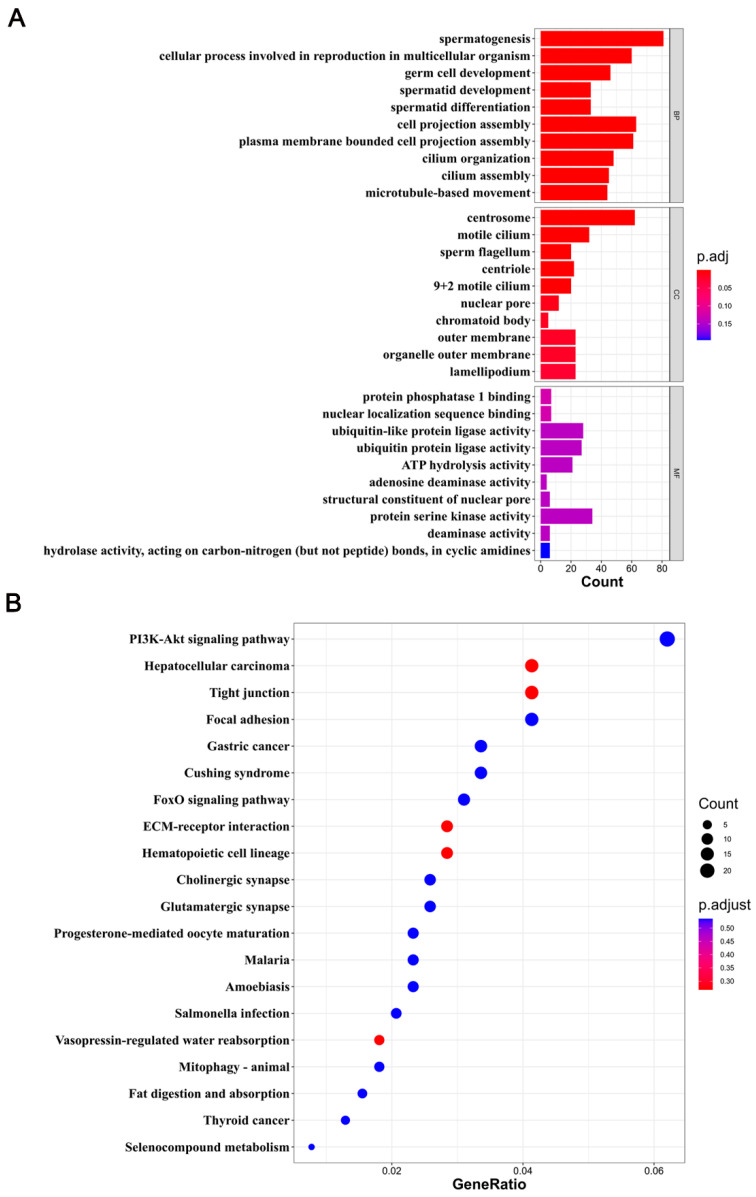
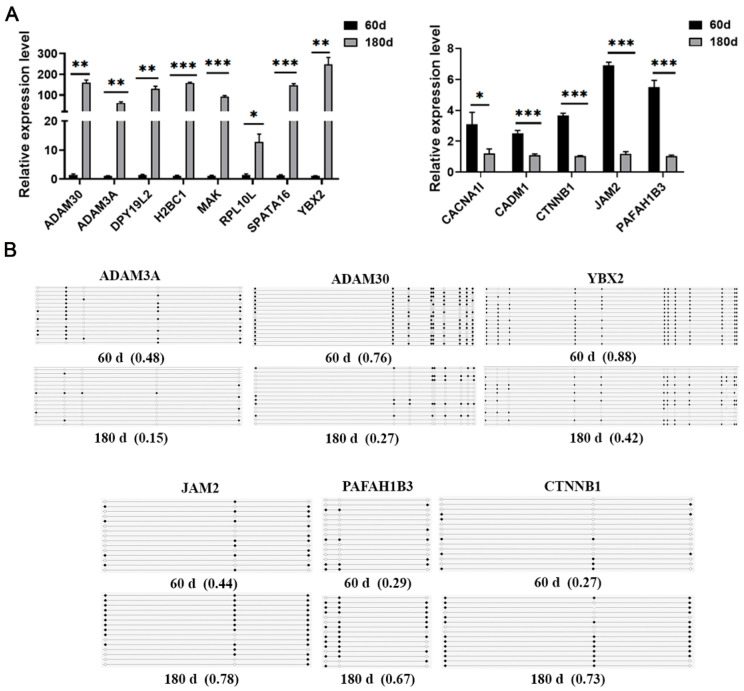
DNA methylation plays a critical role in regulating gene expression during testicular development. However, few studies report on candidate genes related to the DNA methylation regulation of porcine testicular development. This study examined the differentially expressed genes (DEGs) and their methylation levels in testicular tissues from pigs at 60 days of age (60 d) and 180 days of age (180 d) using RNA-Seq and whole genome bisulfite sequencing (WGBS). It was determined that DNA methylation primarily occurs in the cytosine-guanine (CG) context, and the analysis identified 106,282 differentially methylated regions (DMRs) corresponding to 12,385 differentially methylated genes (DMGs). Further integrated analysis of RNA-Seq and WGBS data revealed 1083 DMGs negatively correlated with the expression of DEGs. GO analysis showed that these genes were significantly enriched in spermatogenesis, germ cell development, and spermatid differentiation. The screening of enriched genes revealed that hyper-methylation repressed ADAM30, ADAM3A, DPY19L2, H2BC1, MAK, RPL10L, SPATA16, and YBX2, while hypo-methylation elevated CACNA1I, CADM1, CTNNB1, JAM2, and PAFAH1B3 expression. Additionally, the methylation status of the key genes ADAM3A, ADAM30, YBX2, JAM2, PAFAH1B3, and CTNNB1 was detected by bisulfite sequencing PCR (BSP). This study offers insights into the epigenetic regulation mechanisms underlying porcine testicular development.
Keywords: DNA methylation; RNA-Seq; WGBS; porcine; testicular development.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Liu M., Xu Q., Zhao J., Guo Y., Zhang C., Chao X., Cheng M., Schinckel A.P., Zhou B. Comprehensive Transcriptome Analysis of Follicles from Two Stages of the Estrus Cycle of Two Breeds Reveals the Roles of Long Intergenic Non-Coding RNAs in Gilts. Biology. 2022;11:716. doi: 10.3390/biology11050716. - DOI - PMC - PubMed
MeSH terms
Grants and funding
- 2023M731039/China Postdoctoral Science Foundation
- 2022021302024853/Wuhan Science and Technology Major Project on Key techniques of Biological Breeding and Breeding of New Varieties
- 32102527/National Natural Science Foundation of China
- 2022ABA002/Hubei Provincial Science and Technology Major Project of China
- CARS-35/National Pig Industry Technology System
LinkOut - more resources
Full Text Sources
Miscellaneous
