

Pediatric Gaucher Disease Type 3 Presenting with Oculomotor Apraxia: A Case Report
- PMID: 39201895
- PMCID: PMC11352849
- DOI: 10.3390/children11080960
Pediatric Gaucher Disease Type 3 Presenting with Oculomotor Apraxia: A Case Report
Abstract
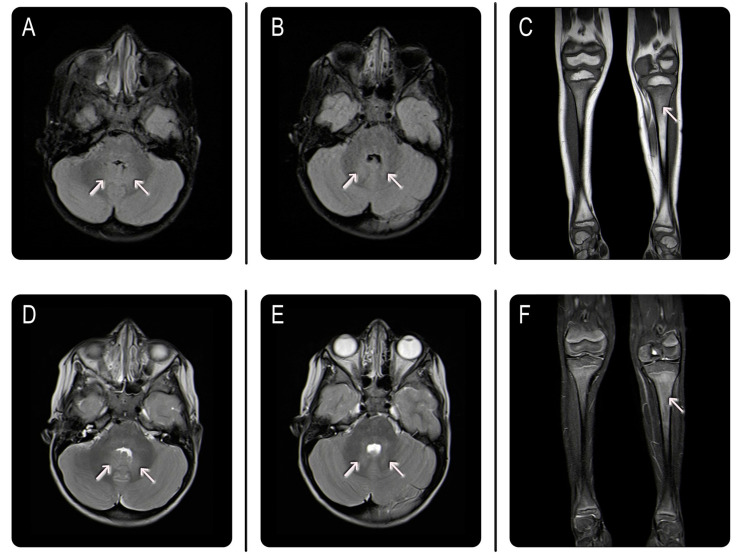
We report on a 4-year-old boy affected by Gaucher disease (GD) type 3, who presented with splenomegaly and a history of oculomotor apraxia. GD is a rare lysosomal storage disorder caused by glucocerebrosidase deficiency with multi-organ involvement. Besides common clinical features such as hepatosplenomegaly and skeletal involvement, less frequent neurological symptoms, such as oculomotor apraxia, are indicative of neuronopathic forms of the disease, namely GD type 3, to be confirmed both by enzyme activity and genetic testing. Overall, GD management requires a multidisciplinary approach involving metabolic pediatricians, neurologists, psychologists, and geneticists, and currently relies on early enzyme replacement therapy. Although enzyme replacement therapy has proved to be effective in improving systemic signs and symptoms, it is unable to alleviate neurological complications once these have occurred, as it does not pass across the blood-brain barrier. Neurological improvements may occur through indirect mechanisms. Thus, our case report aims to highlight the importance of considering GD in the differential diagnosis of pediatric patients presenting with splenomegaly associated with neurological manifestations, as early intervention may significantly modify the disease progression and prevent further irreversible complications.
Keywords: beta-glucocerebrosidase; enzyme replacement therapy; inherited metabolic diseases; lysosomal storage disorder; splenomegaly.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Weinreb N.J., Goker-Alpan O., Kishnani P.S., Longo N., Burrow T.A., Bernat J.A., Gupta P., Henderson N., Pedro H., Prada C.E., et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol. Genet. Metab. 2022;136:4–21. doi: 10.1016/j.ymgme.2022.03.001. - DOI - PubMed
-
- Stirnemann J., Belmatoug N., Camou F., Serratrice C., Froissart R., Caillaud C., Levade T., Astudillo L., Serratrice J., Brassier A., et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017;18:441. doi: 10.3390/ijms18020441. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
