

Integrative Analysis of Multi-Omics Data to Identify Deregulated Molecular Pathways and Druggable Targets in Chronic Lymphocytic Leukemia
- PMID: 39202022
- PMCID: PMC11355716
- DOI: 10.3390/jpm14080831
Integrative Analysis of Multi-Omics Data to Identify Deregulated Molecular Pathways and Druggable Targets in Chronic Lymphocytic Leukemia
Abstract
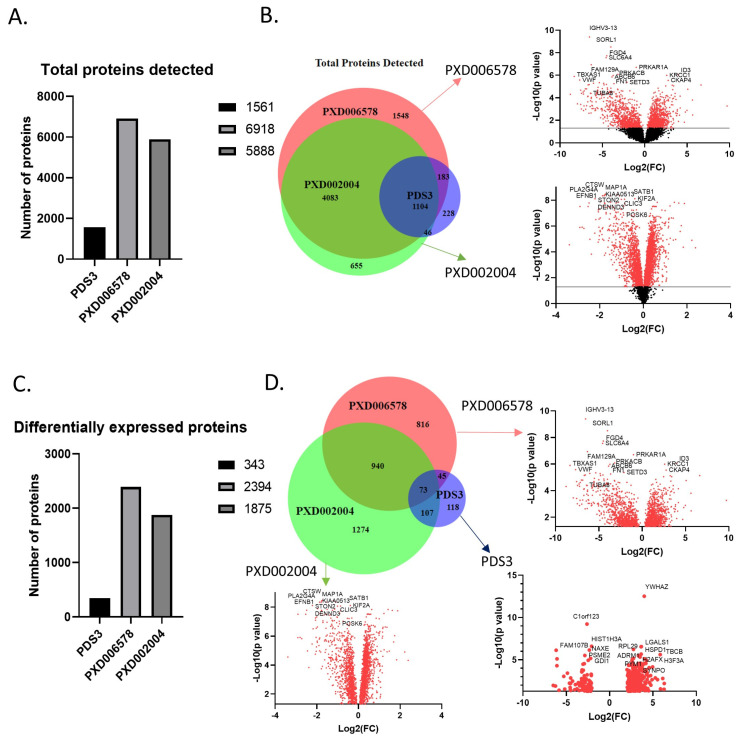
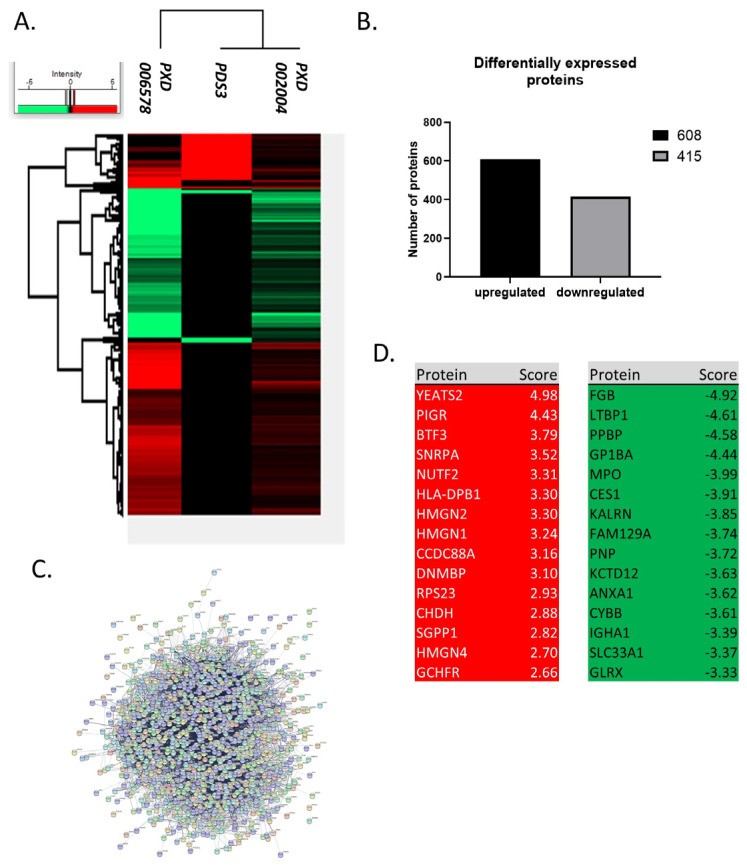
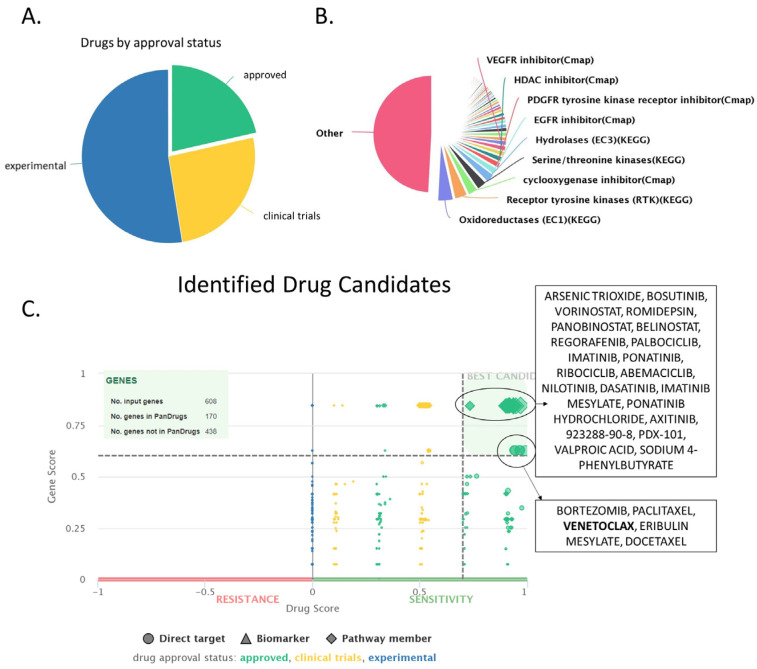
Chronic Lymphocytic Leukemia (CLL) is the most common B-cell malignancy in the Western world, characterized by frequent relapses despite temporary remissions. Our study integrated publicly available proteomic, transcriptomic, and patient survival datasets to identify key differences between healthy and CLL samples. We exposed approximately 1000 proteins that differentiate healthy from cancerous cells, with 608 upregulated and 415 downregulated in CLL cases. Notable upregulated proteins include YEATS2 (an epigenetic regulator), PIGR (Polymeric immunoglobulin receptor), and SNRPA (a splicing factor), which may serve as prognostic biomarkers for this disease. Key pathways implicated in CLL progression involve RNA processing, stress resistance, and immune response deficits. Furthermore, we identified three existing drugs-Bosutinib, Vorinostat, and Panobinostat-for potential further investigation in drug repurposing in CLL. We also found limited correlation between transcriptomic and proteomic data, emphasizing the importance of proteomics in understanding gene expression regulation mechanisms. This generally known disparity highlights once again that mRNA levels do not accurately predict protein abundance due to many regulatory factors, such as protein degradation, post-transcriptional modifications, and differing rates of translation. These results demonstrate the value of integrating omics data to uncover deregulated proteins and pathways in cancer and suggest new therapeutic avenues for CLL.
Keywords: chronic lymphocytic leukemia; drug repurposing; lymphoma; personalized medicine; proteomics; transcriptomics.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures







References
LinkOut - more resources
Full Text Sources
Miscellaneous
