

Recent Progress in Gene-Targeting Therapies for Spinal Muscular Atrophy: Promises and Challenges
- PMID: 39202360
- PMCID: PMC11353366
- DOI: 10.3390/genes15080999
Recent Progress in Gene-Targeting Therapies for Spinal Muscular Atrophy: Promises and Challenges
Abstract
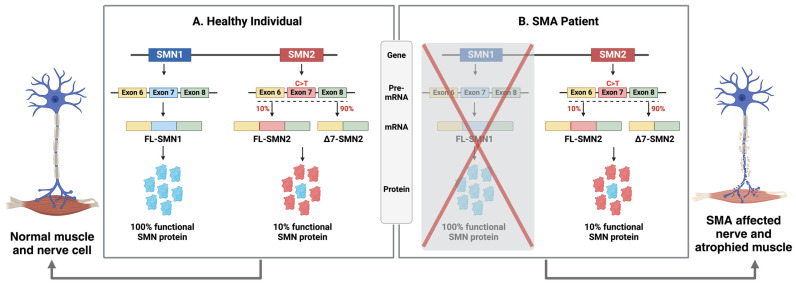
Spinal muscular atrophy (SMA) is a severe genetic disorder characterized by the loss of motor neurons, leading to progressive muscle weakness, loss of mobility, and respiratory complications. In its most severe forms, SMA can result in death within the first two years of life if untreated. The condition arises from mutations in the SMN1 (survival of motor neuron 1) gene, causing a deficiency in the survival motor neuron (SMN) protein. Humans possess a near-identical gene, SMN2, which modifies disease severity and is a primary target for therapies. Recent therapeutic advancements include antisense oligonucleotides (ASOs), small molecules targeting SMN2, and virus-mediated gene replacement therapy delivering a functional copy of SMN1. Additionally, recognizing SMA's broader phenotype involving multiple organs has led to the development of SMN-independent therapies. Evidence now indicates that SMA affects multiple organ systems, suggesting the need for SMN-independent treatments along with SMN-targeting therapies. No single therapy can cure SMA; thus, combination therapies may be essential for comprehensive treatment. This review addresses the SMA etiology, the role of SMN, and provides an overview of the rapidly evolving therapeutic landscape, highlighting current achievements and future directions.
Keywords: SMN protein; SMN2; antisense oligonucleotide (ASO); combination therapy; gene therapy; nusinersen; onasemnogene; risdiplam; small molecule; spinal muscular atrophy (SMA); survival of motor neuron 1 (SMN1).
Conflict of interest statement
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand?Int J Mol Sci. 2020 May 7;21(9):3297. doi: 10.3390/ijms21093297. Int J Mol Sci. 2020. PMID: 32392694 Free PMC article. Review.
-
Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy.Cells. 2022 Jan 26;11(3):417. doi: 10.3390/cells11030417. Cells. 2022. PMID: 35159227 Free PMC article. Review.
-
Recent Advance in Disease Modifying Therapies for Spinal Muscular Atrophy.Acta Neurol Taiwan. 2024 Sep 30;33(3):81-88. Acta Neurol Taiwan. 2024. PMID: 39363429 Review.
-
An Overview of the Therapeutic Strategies for the Treatment of Spinal Muscular Atrophy.Hum Gene Ther. 2023 Mar;34(5-6):180-191. doi: 10.1089/hum.2022.189. Hum Gene Ther. 2023. PMID: 36762938 Review.
-
Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape.Hum Mol Genet. 2017 Oct 1;26(R2):R151-R159. doi: 10.1093/hmg/ddx215. Hum Mol Genet. 2017. PMID: 28977438 Review.
Cited by
-
Hearing loss: a global view for gene therapy approaches and challenges.Eur J Pediatr. 2025 Aug 27;184(9):578. doi: 10.1007/s00431-025-06426-9. Eur J Pediatr. 2025. PMID: 40858759 Free PMC article. Review.
-
Discovery of RNA-Targeting Small Molecules: Challenges and Future Directions.MedComm (2020). 2025 Aug 24;6(9):e70342. doi: 10.1002/mco2.70342. eCollection 2025 Sep. MedComm (2020). 2025. PMID: 40859960 Free PMC article. Review.
-
Advancing personalized spinal muscular atrophy care: matching the right biomarker to the right patient at the right time.J Neurol. 2025 Sep 2;272(9):605. doi: 10.1007/s00415-025-13314-7. J Neurol. 2025. PMID: 40892138 Free PMC article. Review.
-
Recent Progress of Antisense Oligonucleotide Therapy for Superoxide-Dismutase-1-Mutated Amyotrophic Lateral Sclerosis: Focus on Tofersen.Genes (Basel). 2024 Oct 20;15(10):1342. doi: 10.3390/genes15101342. Genes (Basel). 2024. PMID: 39457466 Free PMC article. Review.
-
[Research progress on phenotypic modifier genes in spinal muscular atrophy].Zhongguo Dang Dai Er Ke Za Zhi. 2025 Feb 15;27(2):229-235. doi: 10.7499/j.issn.1008-8830.2410064. Zhongguo Dang Dai Er Ke Za Zhi. 2025. PMID: 39962788 Free PMC article. Review. Chinese.
References
-
- Schrank B., Gotz R., Gunnersen J.M., Ure J.M., Toyka K.V., Smith A.G., Sendtner M. Inactivation of the Survival Motor Neuron Gene, a Candidate Gene for Human Spinal Muscular Atrophy, Leads to Massive Cell Death in Early Mouse Embryos. Proc. Natl. Acad. Sci. USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
