

Nuclear proteasomes buffer cytoplasmic proteins during autophagy compromise
- PMID: 39209961
- PMCID: PMC11469956
- DOI: 10.1038/s41556-024-01488-7
Nuclear proteasomes buffer cytoplasmic proteins during autophagy compromise
Abstract
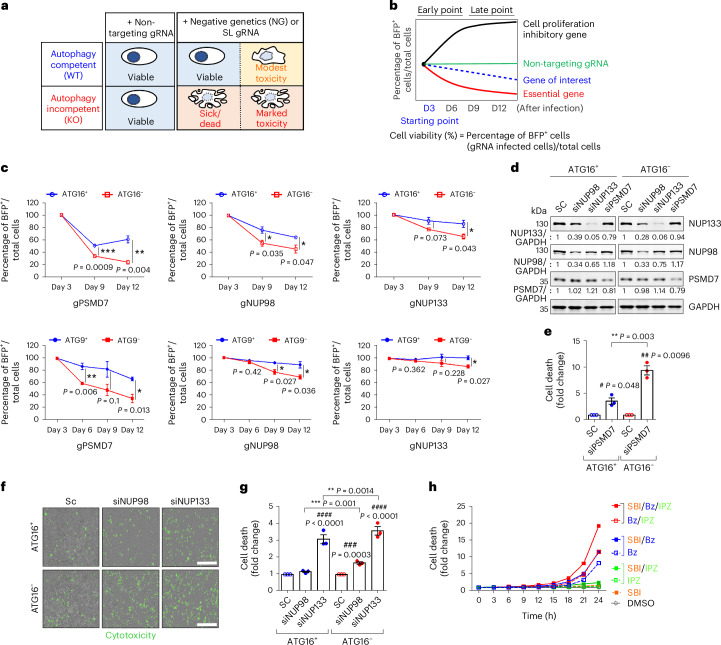
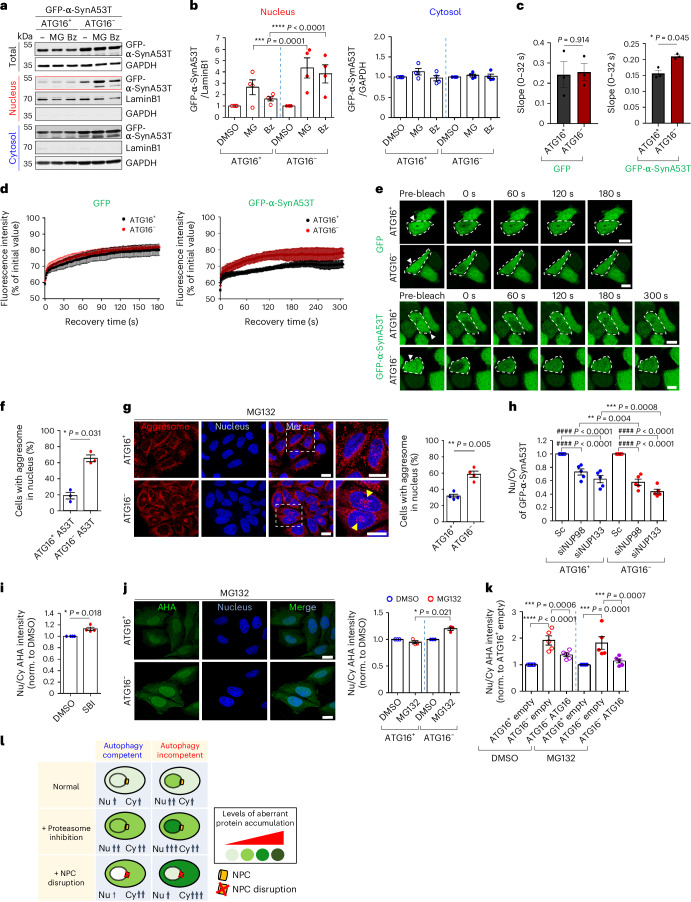
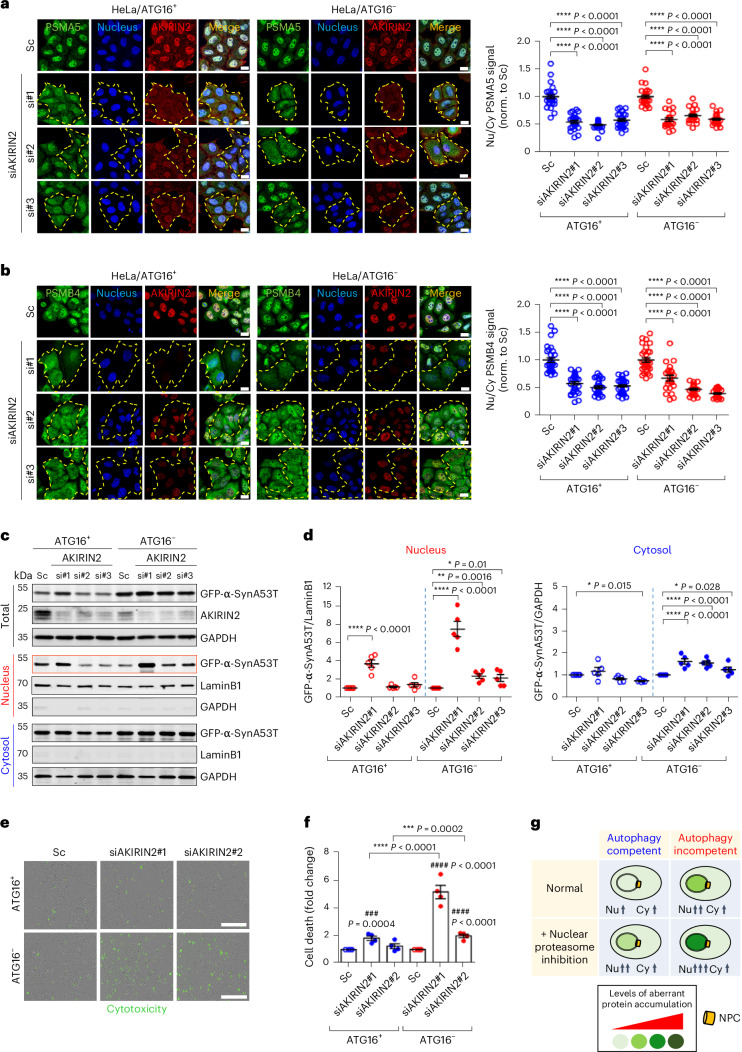
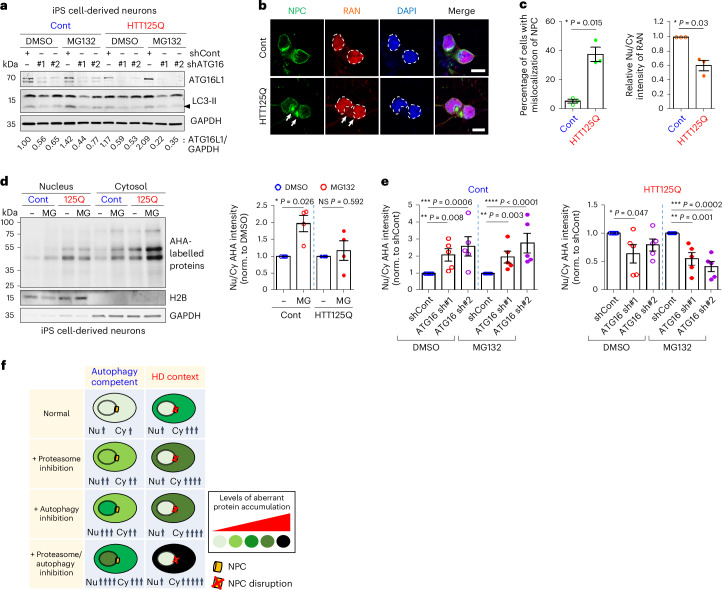
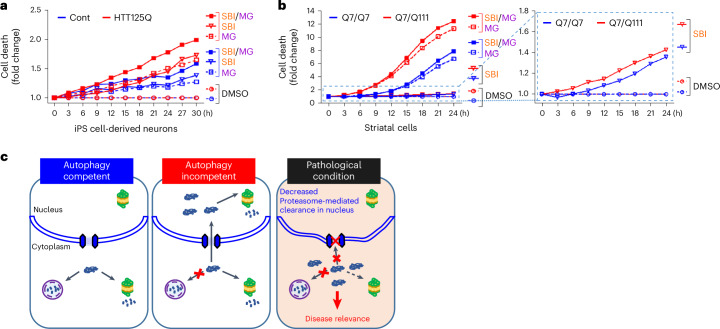
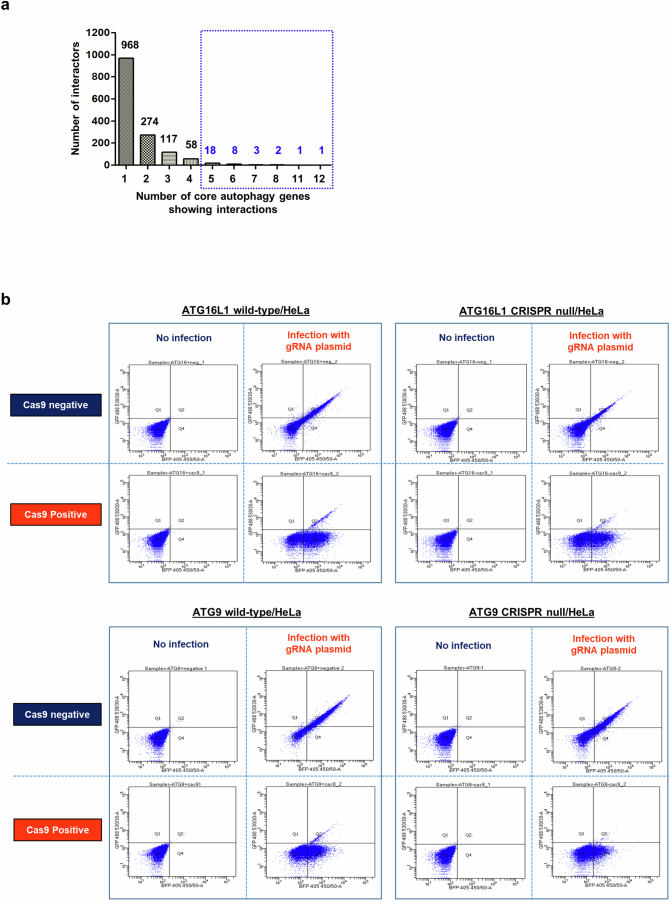
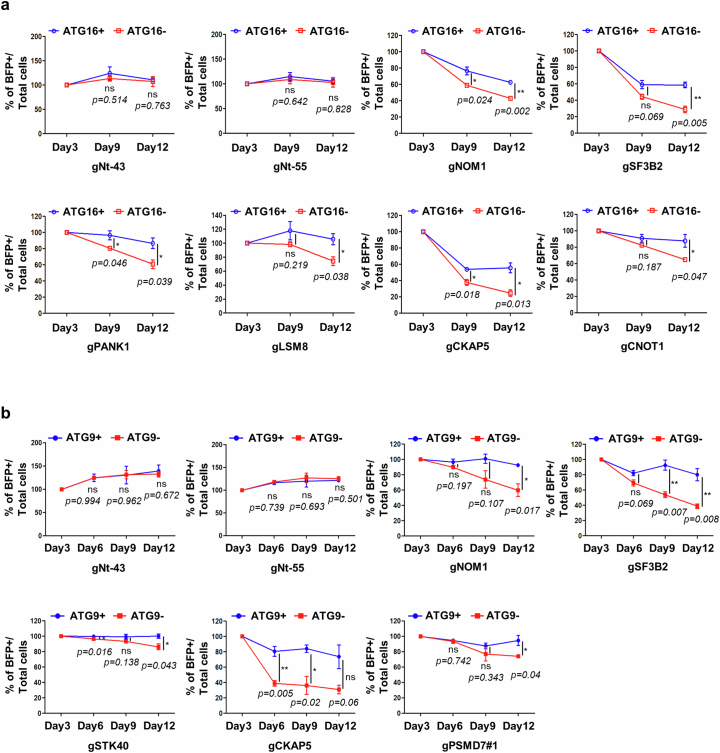
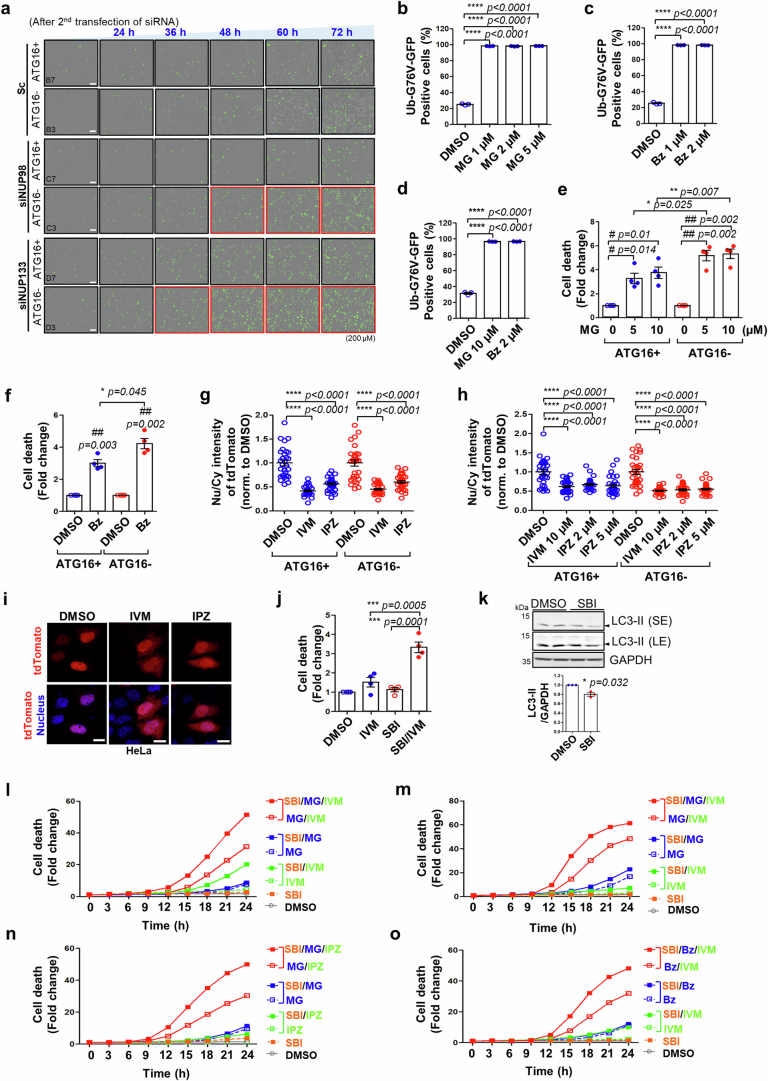
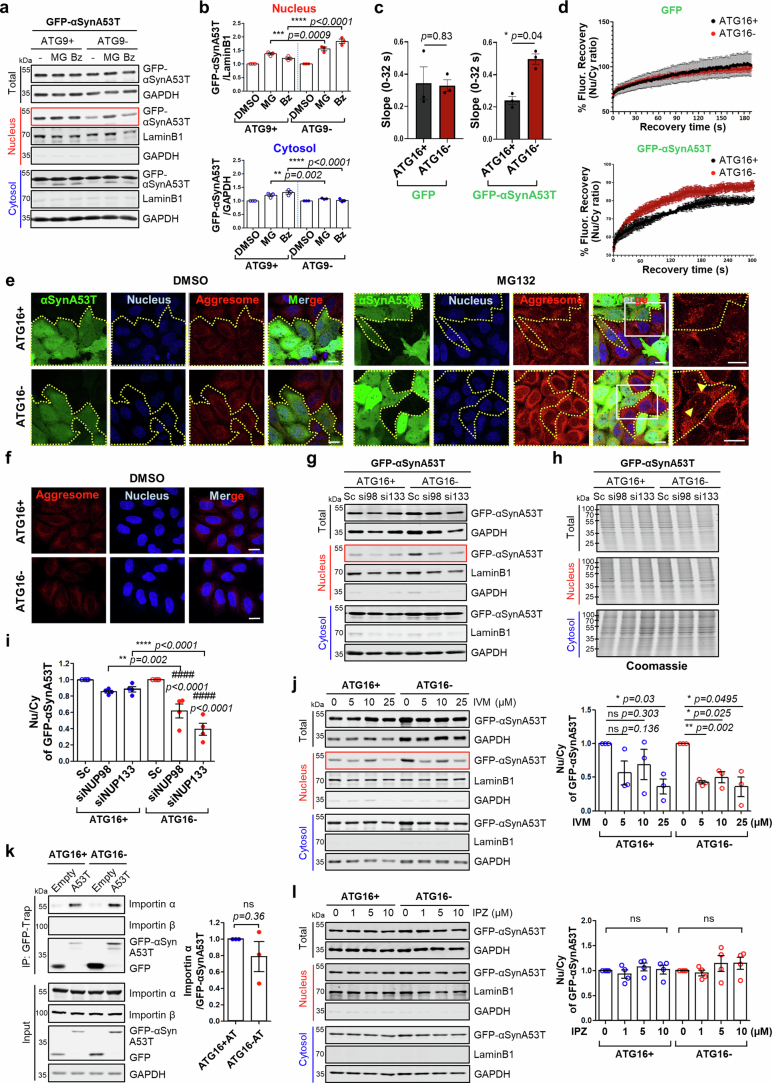
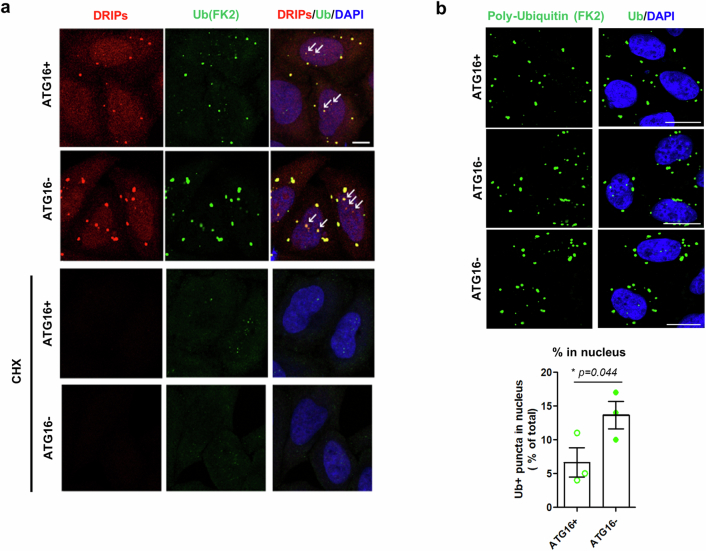
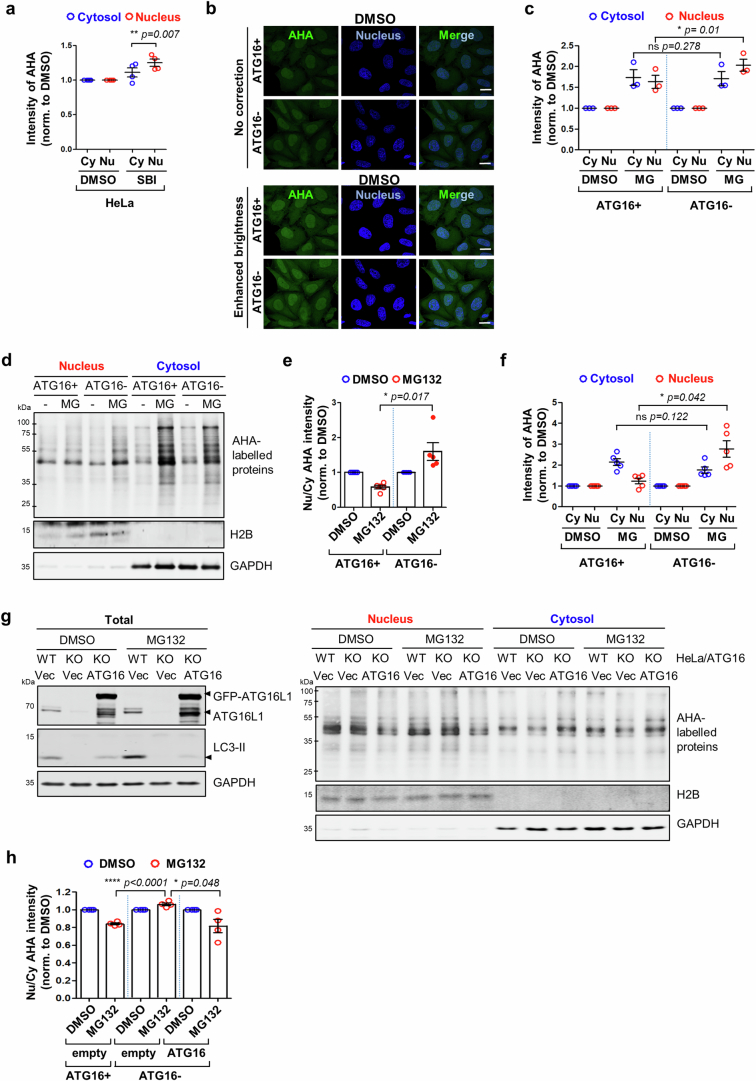
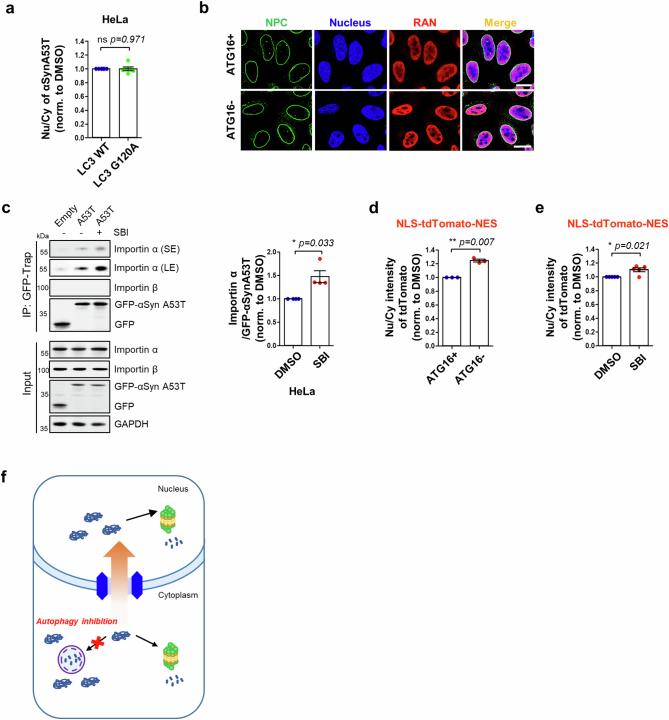
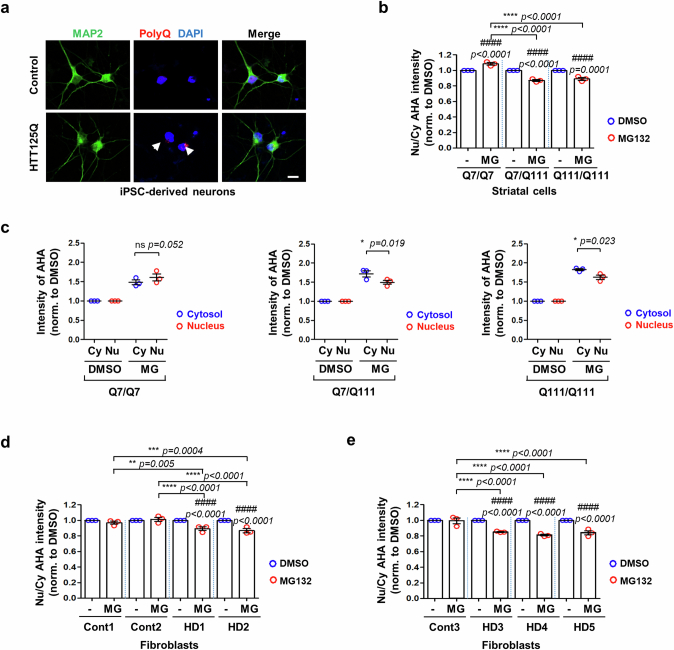
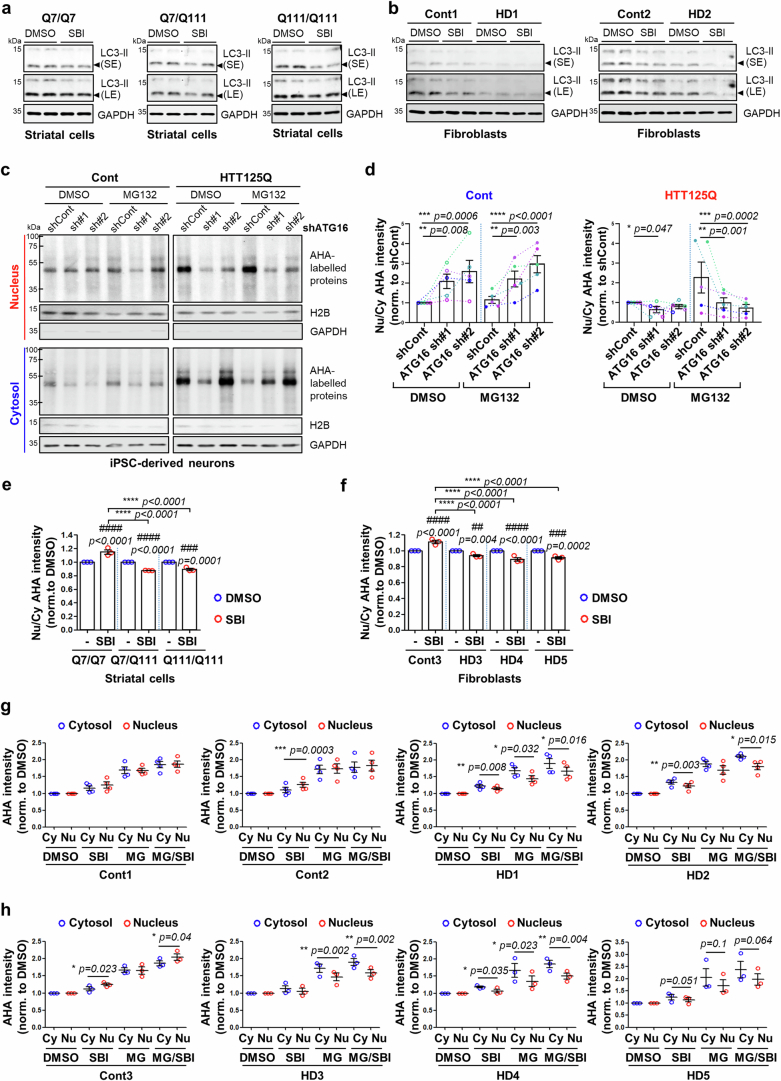
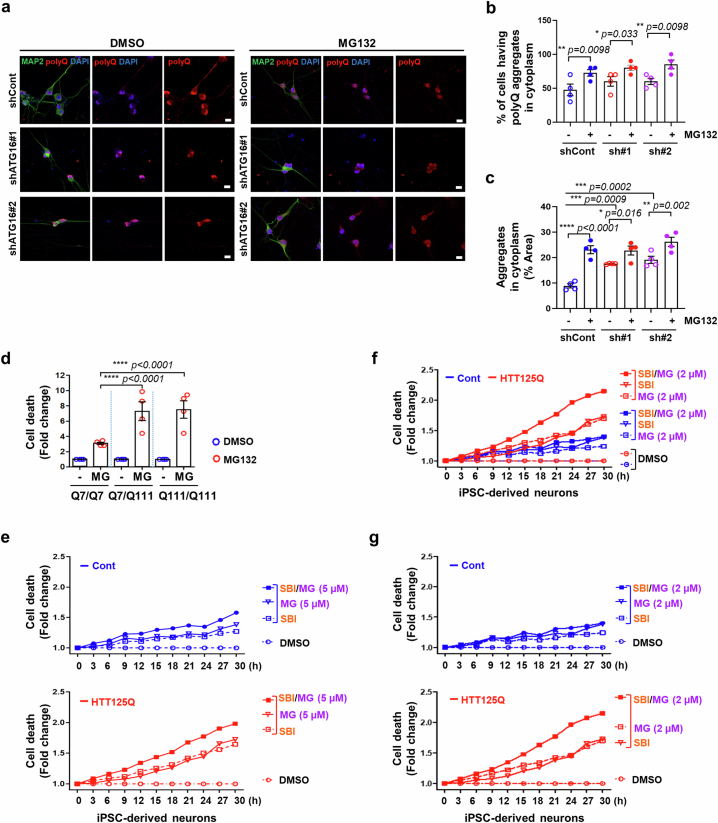
Autophagy is a conserved pathway where cytoplasmic contents are engulfed by autophagosomes, which then fuse with lysosomes enabling their degradation. Mutations in core autophagy genes cause neurological conditions, and autophagy defects are seen in neurodegenerative diseases such as Parkinson's disease and Huntington's disease. Thus, we have sought to understand the cellular pathway perturbations that autophagy-perturbed cells are vulnerable to by seeking negative genetic interactions such as synthetic lethality in autophagy-null human cells using available data from yeast screens. These revealed that loss of proteasome and nuclear pore complex components cause synergistic viability changes akin to synthetic fitness loss in autophagy-null cells. This can be attributed to the cytoplasm-to-nuclear transport of proteins during autophagy deficiency and subsequent degradation of these erstwhile cytoplasmic proteins by nuclear proteasomes. As both autophagy and cytoplasm-to-nuclear transport are defective in Huntington's disease, such cells are more vulnerable to perturbations of proteostasis due to these synthetic interactions.
© 2024. The Author(s).
Conflict of interest statement
D.C.R. is a consultant for Aladdin Healthcare Technologies, Mindrank AI, Nido Biosciences, Drishti Discoveries, Retro Biosciences, PAQ Therapeutics and Alexion Pharma Intl Ops Limited. G.B. is a founder and Director of Function Rx. The other authors declare no competing interests.
Figures
References
-
- Menzies, F. M., Fleming, A. & Rubinsztein, D. C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci.16, 345–357 (2015). - PubMed
-
- Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet.36, 585–595 (2004). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
