

Advanced sampling simulations of coupled folding and binding of phage P22 N-peptide to boxB RNA
- PMID: 39210596
- PMCID: PMC11480772
- DOI: 10.1016/j.bpj.2024.08.022
Advanced sampling simulations of coupled folding and binding of phage P22 N-peptide to boxB RNA
Abstract
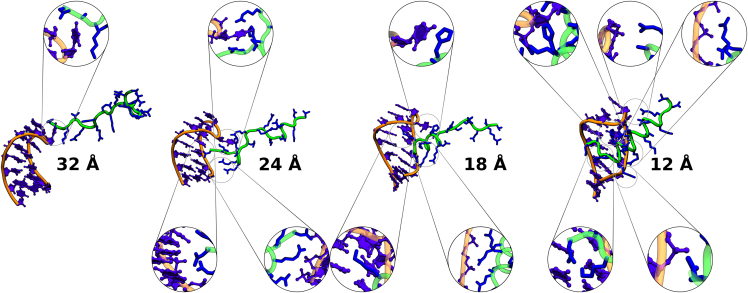
Protein-RNA interactions are crucially important for numerous cellular processes and often involve coupled folding and binding of peptide segments upon association. The Nut-utilization site (N)-protein of bacteriophages contains an N-terminal arginine-rich motif that undergoes such a folding transition upon binding to the boxB RNA hairpin loop target structure. Molecular dynamics free energy simulations were used to calculate the absolute binding free energy of the N-peptide of bacteriophage P22 in complex with the boxB RNA hairpin motif at different salt concentrations and using two different water force field models. We obtained good agreement with experiment also at different salt concentrations for the TIP4P-D water model that has a stabilizing effect on unfolded protein structures. It allowed us to estimate the free energy contribution resulting from restricting the molecules' spatial and conformational freedom upon binding, which makes a large opposing contribution to binding. In a second set of umbrella sampling simulations to dissociate/associate the complex along a separation coordinate, we analyzed the onset of preorientation of the N-peptide and onset of structure formation relative to the RNA and its dependence on the salt concentration. Peptide orientation and conformational transitions are significantly coupled to the first contact formation between peptide and RNA. The initial contacts are mostly formed between peptide residues and the boxB hairpin loop nucleotides. A complete transition to an α-helical bound peptide conformation occurs only at a late stage of the binding process a few angstroms before the complexed state has been reached. However, the N-peptide orients also at distances beyond the contact distance such that the sizable positive charge points toward the RNA's center-of-mass. Our result may have important implications for understanding protein- and peptide-RNA complex formation frequently involving coupled folding and association processes.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures










References
-
- Lund M., Vrbka L., Jungwirth P. Specific Ion Binding to Nonpolar Surface Patches of Proteins. J. Am. Chem. Soc. 2008;130:11582–11583. - PubMed
-
- Patel D.J. Adaptive recognition in RNA complexes with peptides and protein modules. Curr. Opin. Struct. Biol. 1999;9:74–87. - PubMed
-
- Allers J., Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001;311:75–86. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
