

Dominantly acting variants in ATP6V1C1 and ATP6V1B2 cause a multisystem phenotypic spectrum by altering lysosomal and/or autophagosome function
- PMID: 39210597
- PMCID: PMC11465052
- DOI: 10.1016/j.xhgg.2024.100349
Dominantly acting variants in ATP6V1C1 and ATP6V1B2 cause a multisystem phenotypic spectrum by altering lysosomal and/or autophagosome function
Abstract
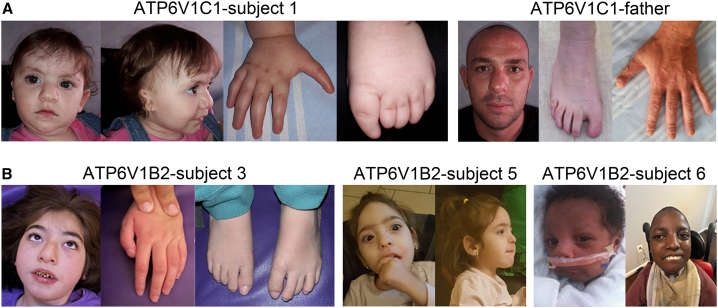
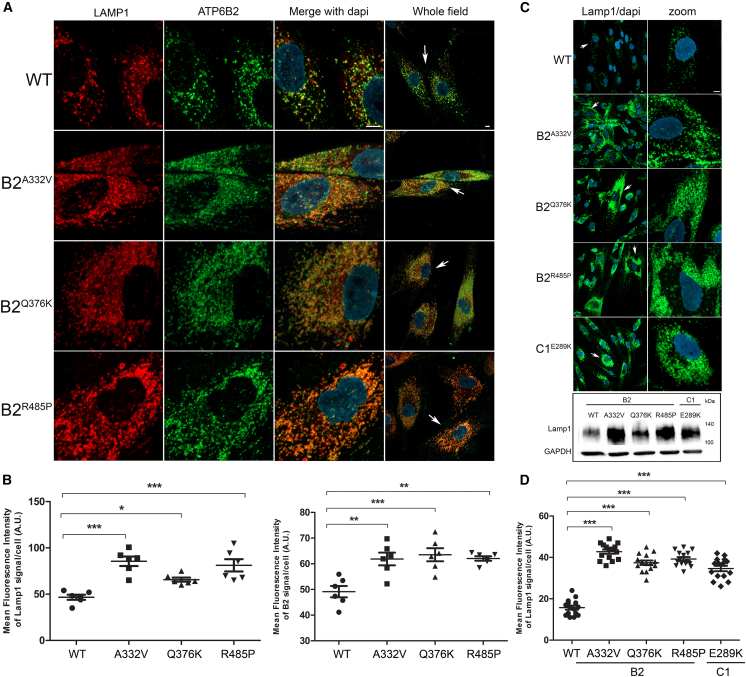
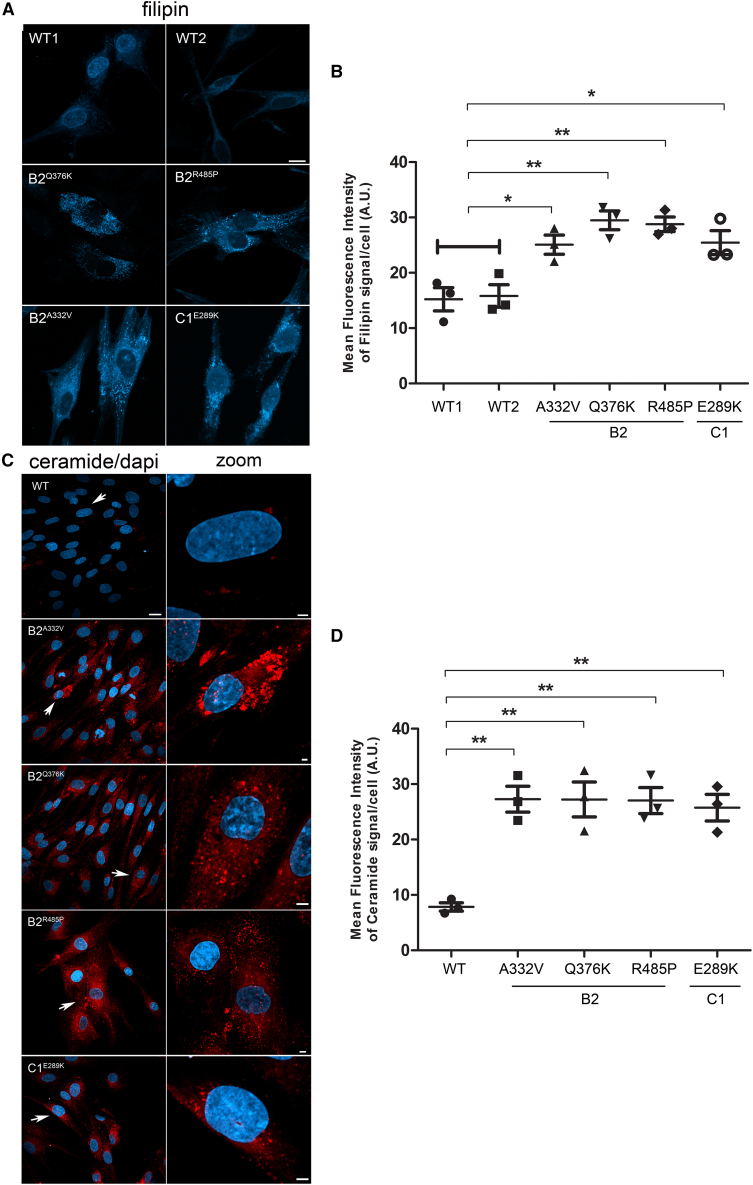
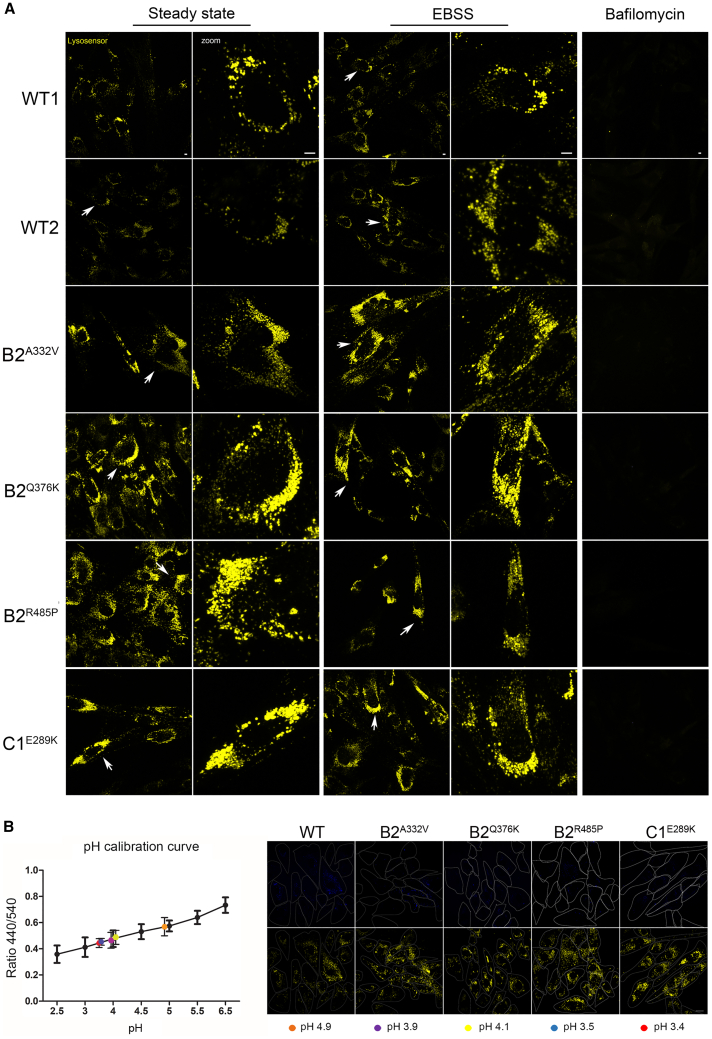
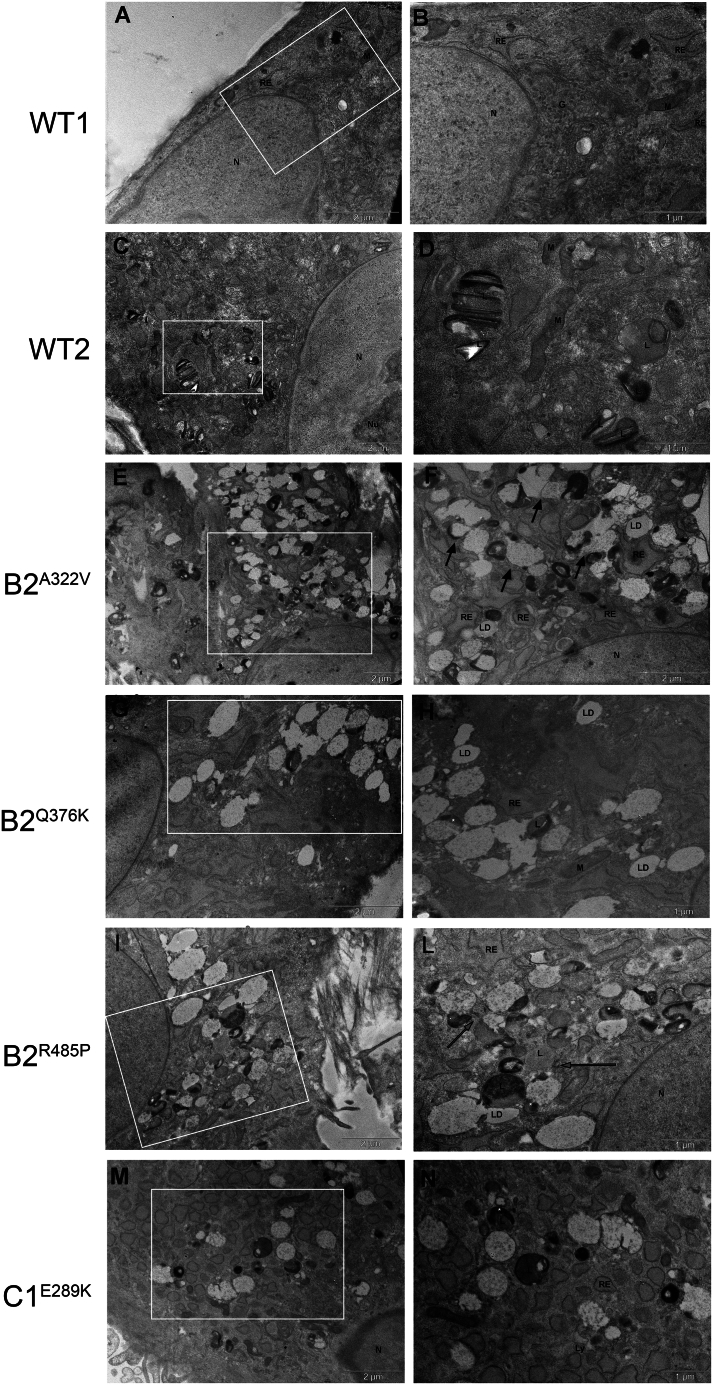
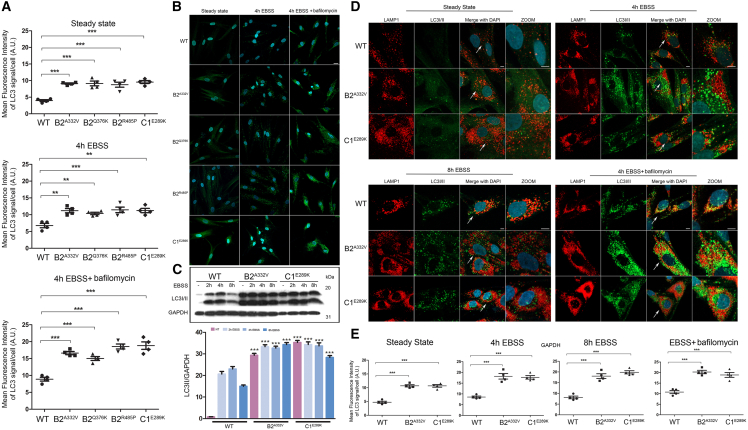
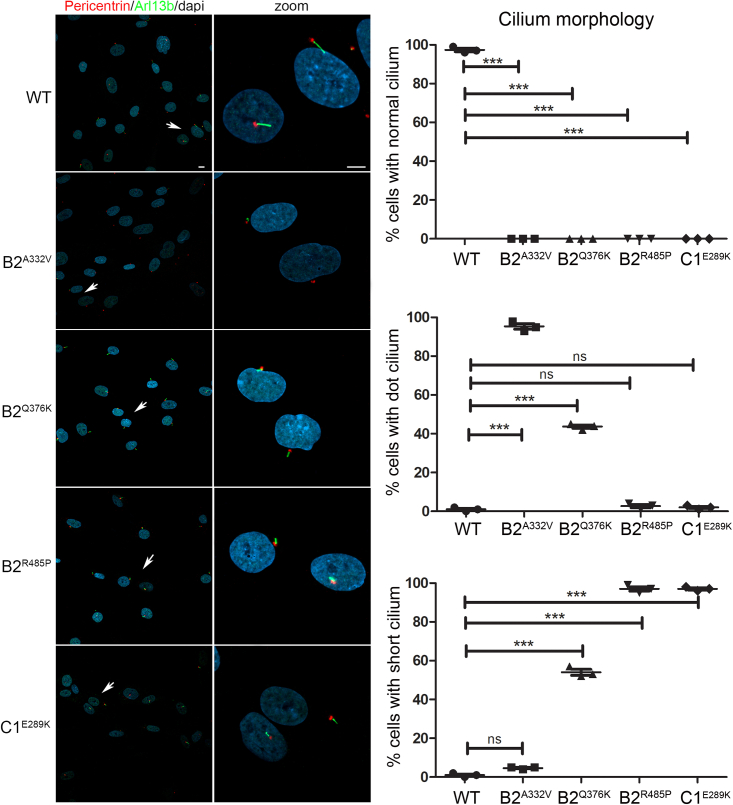
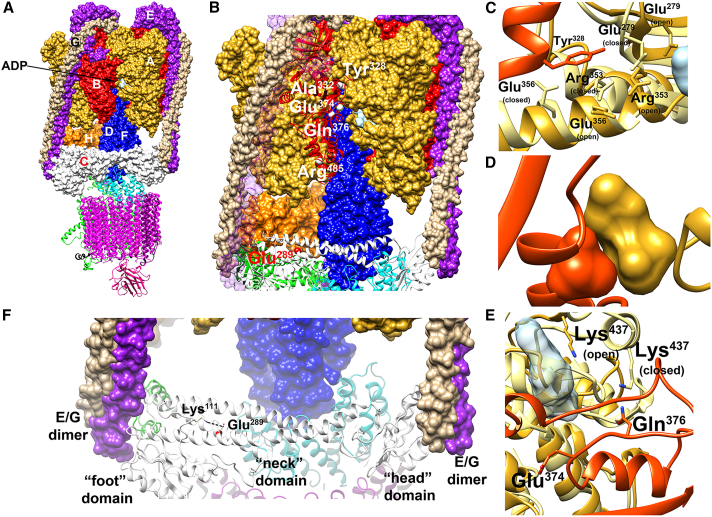
The vacuolar H+-ATPase (V-ATPase) is a functionally conserved multimeric complex localized at the membranes of many organelles where its proton-pumping action is required for proper lumen acidification. The V-ATPase complex is composed of several subunits, some of which have been linked to human disease. We and others previously reported pathogenic dominantly acting variants in ATP6V1B2, the gene encoding the V1B2 subunit, as underlying a clinically variable phenotypic spectrum including dominant deafness-onychodystrophy (DDOD) syndrome, Zimmermann-Laband syndrome (ZLS), and deafness, onychodystrophy, osteodystrophy, intellectual disability, and seizures (DOORS) syndrome. Here, we report on an individual with features fitting DOORS syndrome caused by dysregulated ATP6V1C1 function, expand the clinical features associated with ATP6V1B2 pathogenic variants, and provide evidence that these ATP6V1C1/ATP6V1B2 amino acid substitutions result in a gain-of-function mechanism upregulating V-ATPase function that drives increased lysosomal acidification. We demonstrate a disruptive effect of these ATP6V1B2/ATP6V1C1 variants on lysosomal morphology, localization, and function, resulting in a defective autophagic flux and accumulation of lysosomal substrates. We also show that the upregulated V-ATPase function affects cilium biogenesis, further documenting pleiotropy. This work identifies ATP6V1C1 as a new gene associated with a neurodevelopmental phenotype resembling DOORS syndrome, documents the occurrence of a phenotypic continuum between ZLS, and DDOD and DOORS syndromes, and classify these conditions as lysosomal disorders.
Keywords: ATP6V1B2; ATP6V1C1; DDOD syndrome; DOORS syndrome; Zimmermann-Laband syndrome; autophagy; lysosome; neurodevelopmental disorder; pleiotropy; vacuolar ATPase.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Van Damme T., Gardeitchik T., Mohamed M., Guerrero-Castillo S., Freisinger P., Guillemyn B., Kariminejad A., Dalloyaux D., van Kraaij S., Lefeber D.J., et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. Am. J. Hum. Genet. 2020;107:374. doi: 10.1016/j.ajhg.2020.07.013. - DOI - PMC - PubMed
-
- Bruce L.J., Cope D.L., Jones G.K., Schofield A.E., Burley M., Povey S., Unwin R.J., Wrong O., Tanner M.J. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (band 3, AE1) gene. J. Clin. Invest. 1997;100:1693–1707. doi: 10.1172/JCI119694. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
