

Accelerating drug discovery and repurposing by combining transcriptional signature connectivity with docking
- PMID: 39213358
- PMCID: PMC11364105
- DOI: 10.1126/sciadv.adj3010
Accelerating drug discovery and repurposing by combining transcriptional signature connectivity with docking
Abstract
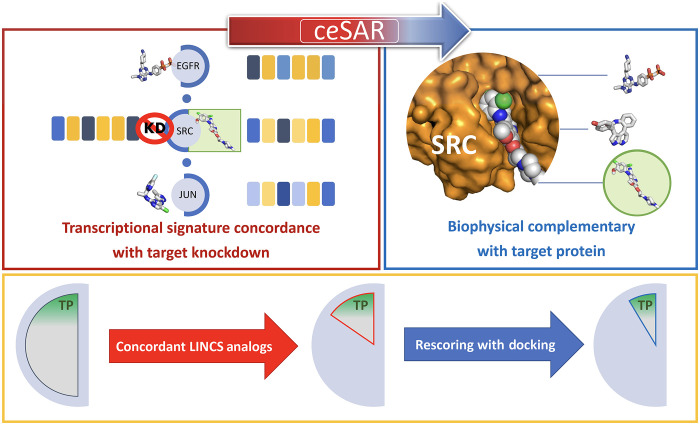
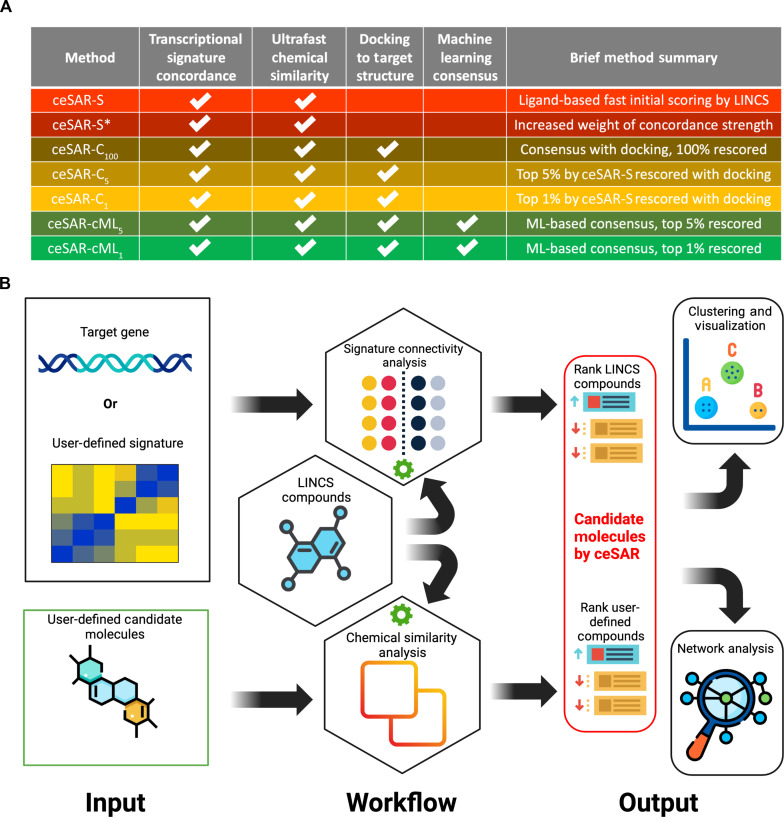
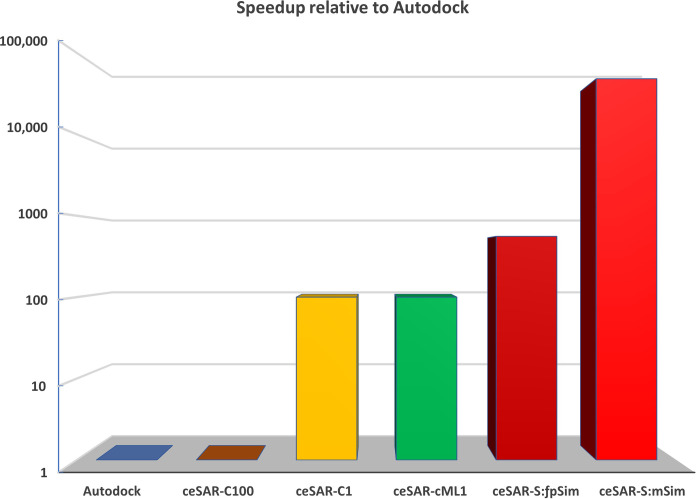
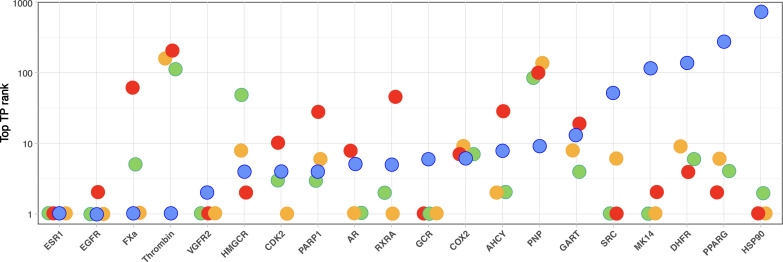
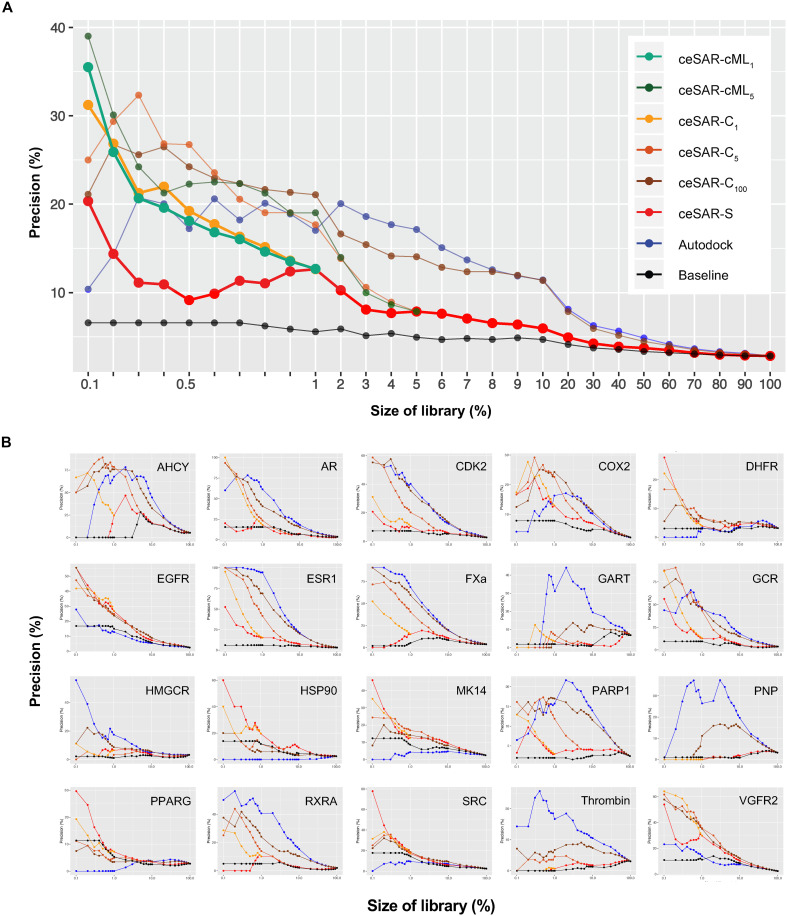
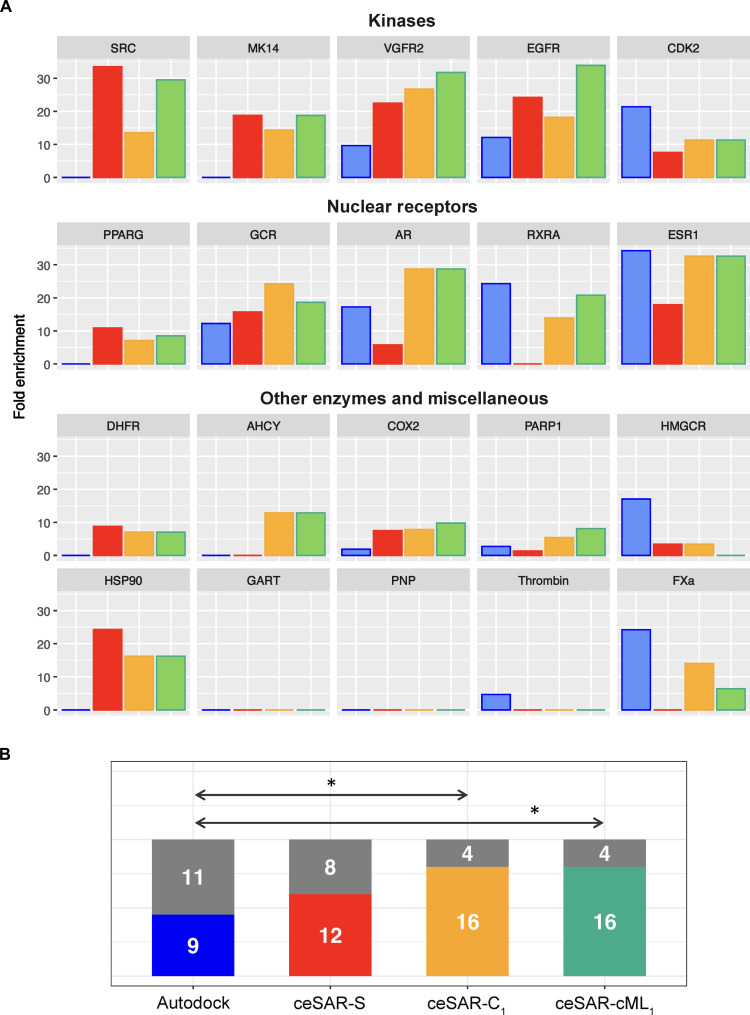
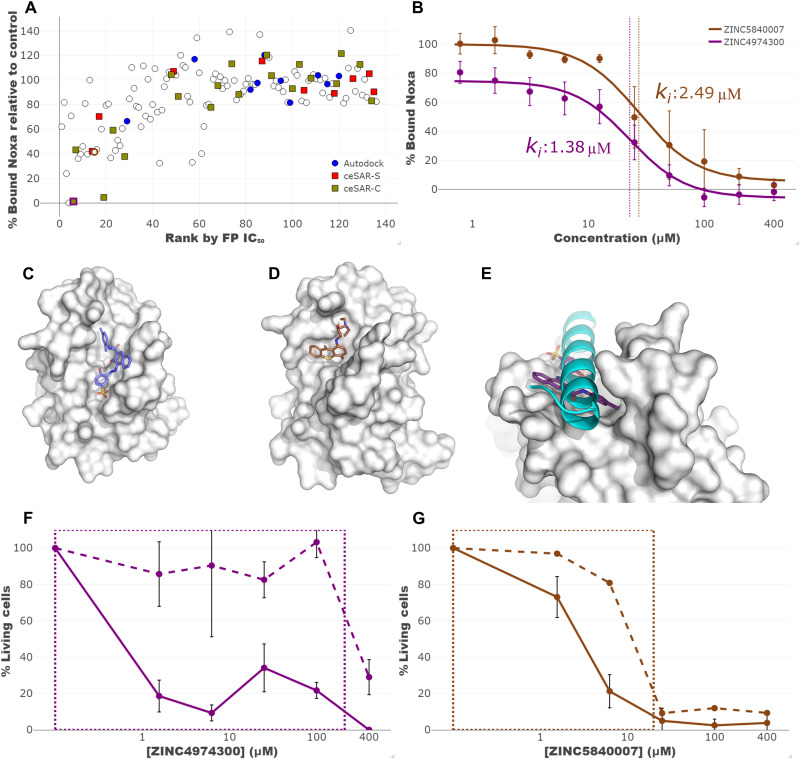
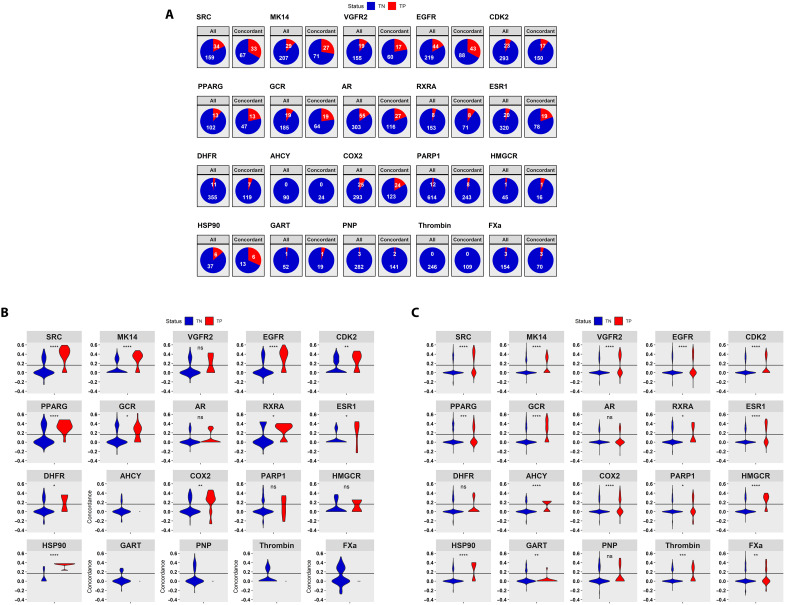
We present an in silico approach for drug discovery, dubbed connectivity enhanced structure activity relationship (ceSAR). Building on the landmark LINCS library of transcriptional signatures of drug-like molecules and gene knockdowns, ceSAR combines cheminformatic techniques with signature concordance analysis to connect small molecules and their targets and further assess their biophysical compatibility using molecular docking. Candidate compounds are first ranked in a target structure-independent manner, using chemical similarity to LINCS analogs that exhibit transcriptomic concordance with a target gene knockdown. Top candidates are subsequently rescored using docking simulations and machine learning-based consensus of the two approaches. Using extensive benchmarking, we show that ceSAR greatly reduces false-positive rates, while cutting run times by multiple orders of magnitude and further democratizing drug discovery pipelines. We further demonstrate the utility of ceSAR by identifying and experimentally validating inhibitors of BCL2A1, an important antiapoptotic target in melanoma and preterm birth-associated inflammation.
Figures
References
-
- Seiler K. P., George G. A., Happ M. P., Bodycombe N. E., Carrinski H. A., Norton S., Brudz S., Sullivan J. P., Muhlich J., Serrano M., Ferraiolo P., Tolliday N. J., Schreiber S. L., Clemons P. A., ChemBank: A small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 36, D351–D359 (2007). - PMC - PubMed
-
- Eder J., Sedrani R., Wiesmann C., The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 13, 577–587 (2014). - PubMed
-
- Lamb J., Crawford E. D., Peck D., Modell J. W., Blat I. C., Wrobel M. J., Lerner J., Brunet J.-P., Subramanian A., Ross K. N., Reich M., Hieronymus H., Wei G., Armstrong S. A., Haggarty S. J., Clemons P. A., Wei R., Carr S. A., Lander E. S., Golub T. R., The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). - PubMed
-
- Shoemaker R. H., Monks A., Alley M. C., Scudiero D. A., Fine D. L., McLemore T. L., Abbott B. J., Paull K. D., Mayo J. G., Boyd M. R., Development of human tumor cell line panels for use in disease-oriented drug screening. Prog. Clin. Biol. Res. 276, 265–286 (1988). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
