

Multi-armored allogeneic MUC1 CAR T cells enhance efficacy and safety in triple-negative breast cancer
- PMID: 39213364
- PMCID: PMC11364110
- DOI: 10.1126/sciadv.adn9857
Multi-armored allogeneic MUC1 CAR T cells enhance efficacy and safety in triple-negative breast cancer
Abstract
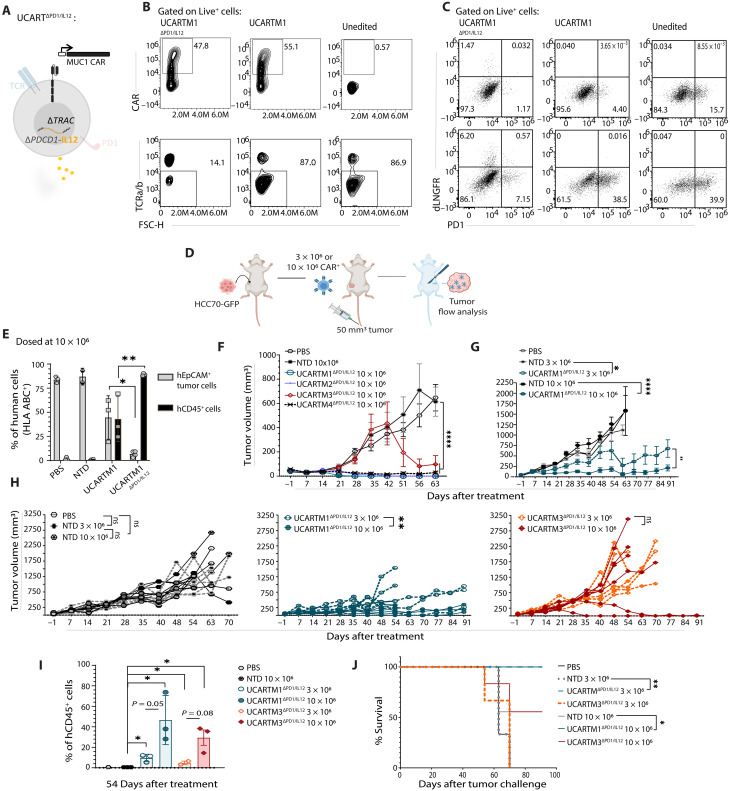
Solid tumors, such as triple-negative breast cancer (TNBC), are biologically complex due to cellular heterogeneity, lack of tumor-specific antigens, and an immunosuppressive tumor microenvironment (TME). These challenges restrain chimeric antigen receptor (CAR) T cell efficacy, underlining the importance of armoring. In solid cancers, a localized tumor mass allows alternative administration routes, such as intratumoral delivery with the potential to improve efficacy and safety but may compromise metastatic-site treatment. Using a multi-layered CAR T cell engineering strategy that allowed a synergy between attributes, we show enhanced cytotoxic activity of MUC1 CAR T cells armored with PD1KO, tumor-specific interleukin-12 release, and TGFBR2KO attributes catered towards the TNBC TME. Intratumoral treatment effectively reduced distant tumors, suggesting retention of antigen-recognition benefits at metastatic sites. Overall, we provide preclinical evidence of armored non-alloreactive MUC1 CAR T cells greatly reducing high TNBC tumor burden in a TGFB1- and PD-L1-rich TME both at local and distant sites while preserving safety.
Figures






References
-
- Harbeck N., Penault-Llorca F., Cortes J., Gnant M., Houssami N., Poortmans P., Ruddy K., Tsang J., Cardoso F., Breast cancer. Nat. Rev. Dis. Primers. 5, 66 (2019). - PubMed
-
- Kumar P., Aggarwal R., An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 293, 247–269 (2016). - PubMed
-
- Bianchini G., De Angelis C., Licata L., Gianni L., Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 19, 91–113 (2022). - PubMed
-
- Dees S., Ganesan R., Singh S., Grewal I. S., Emerging CAR-T cell therapy for the treatment of triple-negative breast cancer. Mol. Cancer Ther. 19, 2409–2421 (2020). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
