

A catalog of small proteins from the global microbiome
- PMID: 39214983
- PMCID: PMC11364881
- DOI: 10.1038/s41467-024-51894-6
A catalog of small proteins from the global microbiome
Abstract
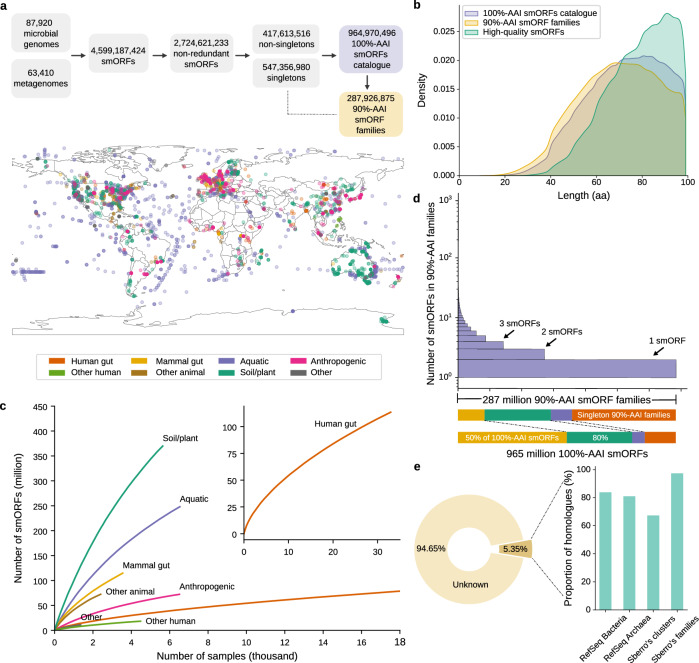
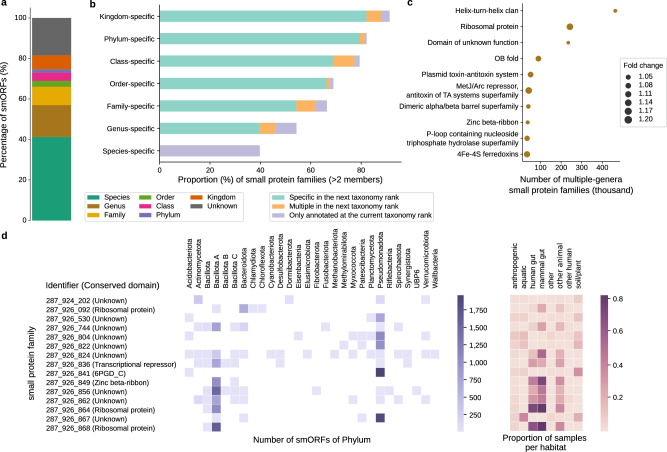
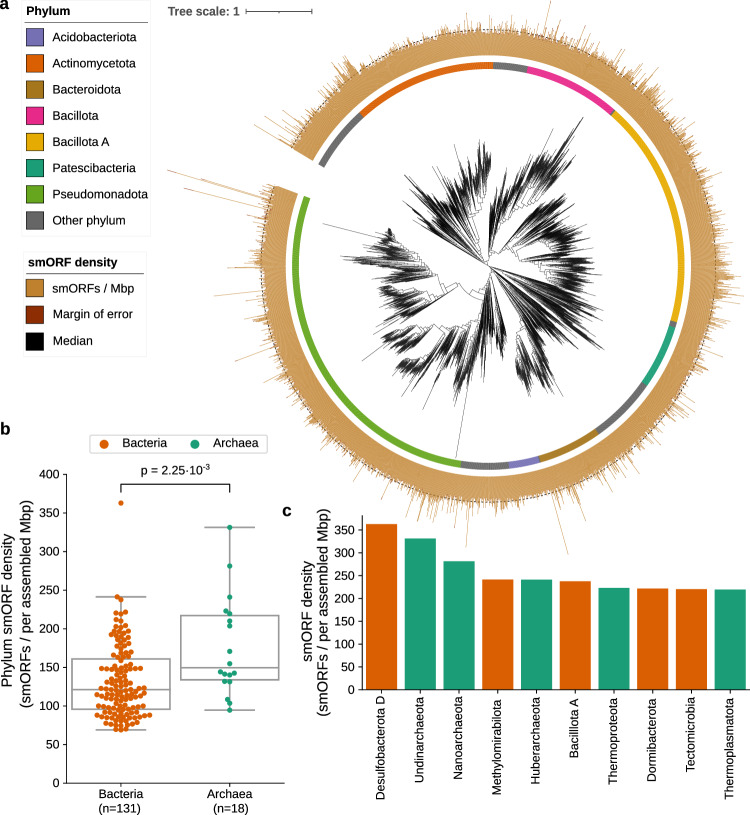
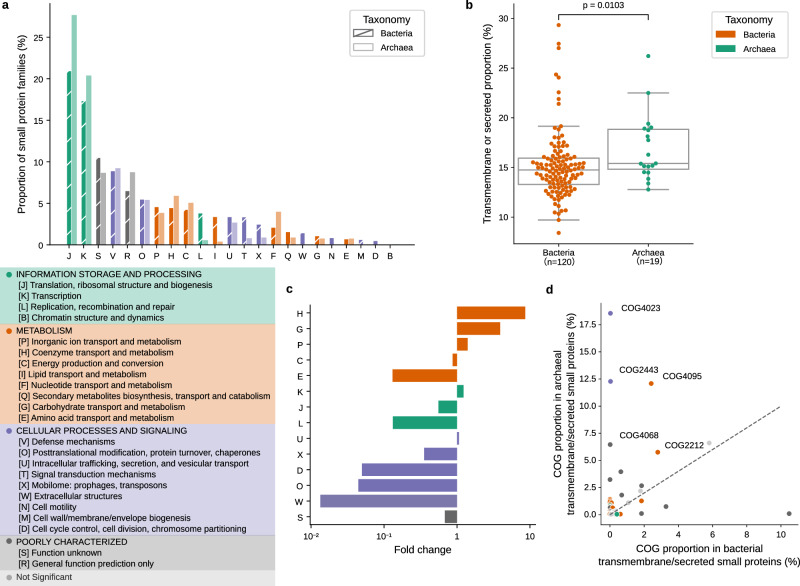
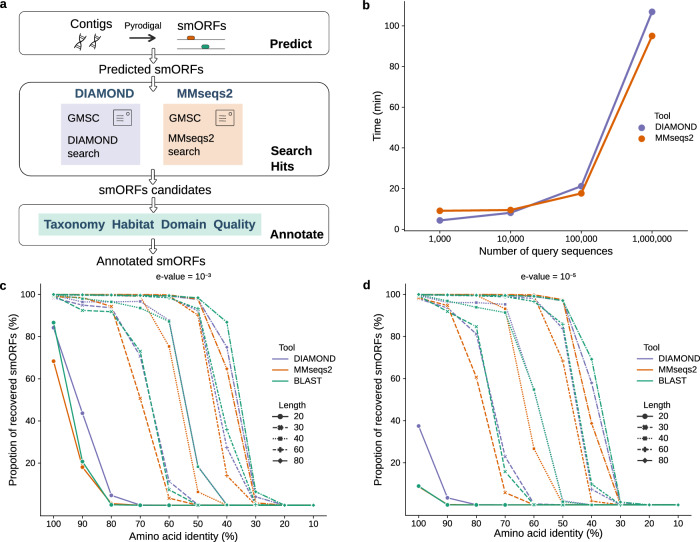
Small open reading frames (smORFs) shorter than 100 codons are widespread and perform essential roles in microorganisms, where they encode proteins active in several cell functions, including signal pathways, stress response, and antibacterial activities. However, the ecology, distribution and role of small proteins in the global microbiome remain unknown. Here, we construct a global microbial smORFs catalog (GMSC) derived from 63,410 publicly available metagenomes across 75 distinct habitats and 87,920 high-quality isolate genomes. GMSC contains 965 million non-redundant smORFs with comprehensive annotations. We find that archaea harbor more smORFs proportionally than bacteria. We moreover provide a tool called GMSC-mapper to identify and annotate small proteins from microbial (meta)genomes. Overall, this publicly-available resource demonstrates the immense and underexplored diversity of small proteins.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
