

DNA methylation governs the sensitivity of repeats to restriction by the HUSH-MORC2 corepressor
- PMID: 39214989
- PMCID: PMC11364546
- DOI: 10.1038/s41467-024-50765-4
DNA methylation governs the sensitivity of repeats to restriction by the HUSH-MORC2 corepressor
Abstract
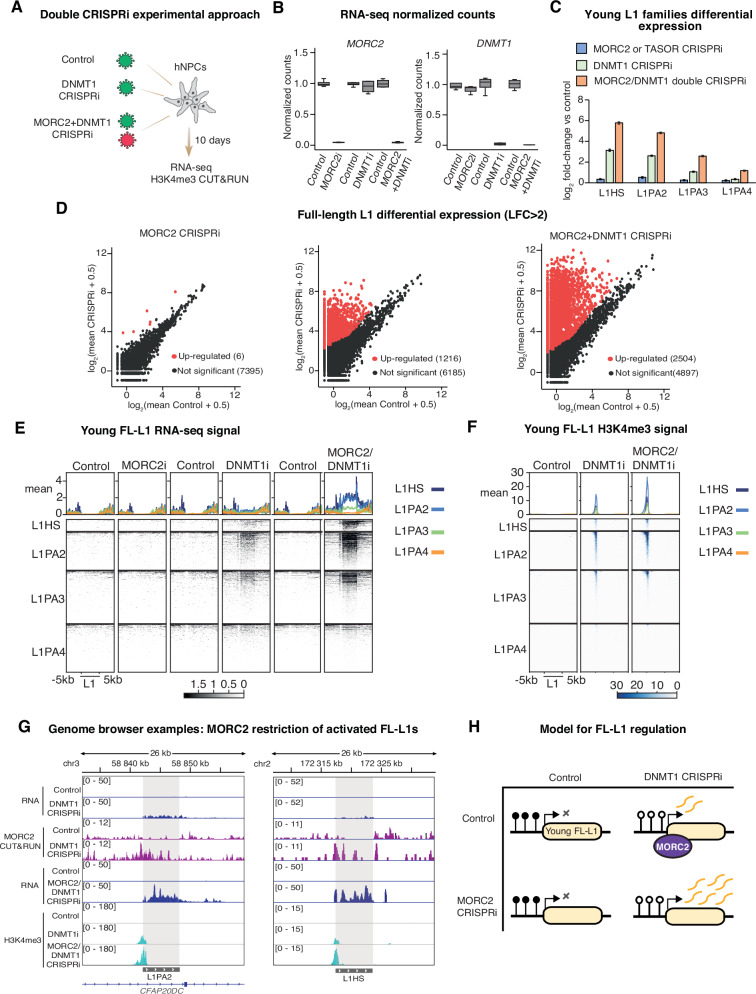
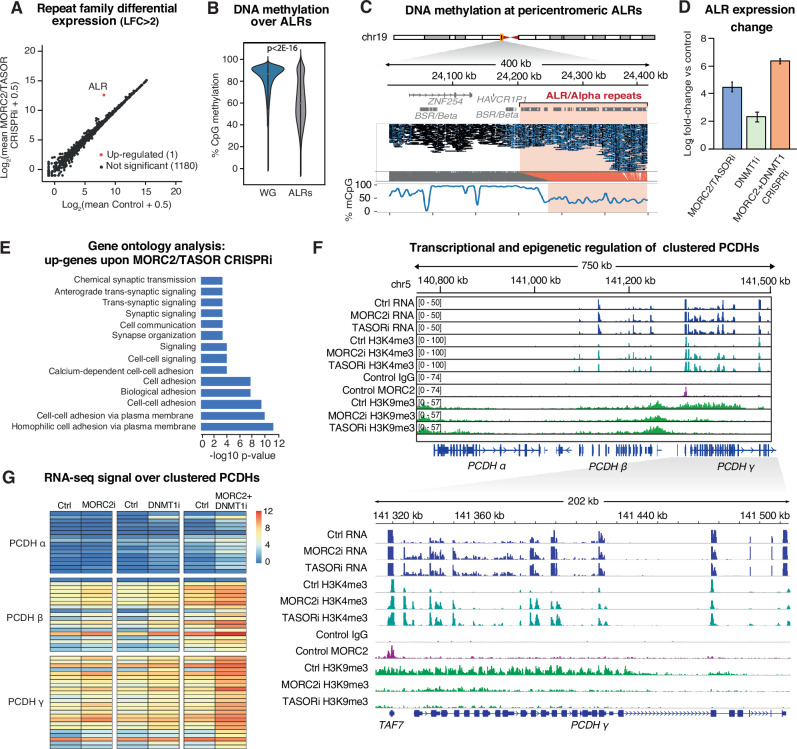
The human silencing hub (HUSH) complex binds to transcripts of LINE-1 retrotransposons (L1s) and other genomic repeats, recruiting MORC2 and other effectors to remodel chromatin. How HUSH and MORC2 operate alongside DNA methylation, a central epigenetic regulator of repeat transcription, remains largely unknown. Here we interrogate this relationship in human neural progenitor cells (hNPCs), a somatic model of brain development that tolerates removal of DNA methyltransferase DNMT1. Upon loss of MORC2 or HUSH subunit TASOR in hNPCs, L1s remain silenced by robust promoter methylation. However, genome demethylation and activation of evolutionarily-young L1s attracts MORC2 binding, and simultaneous depletion of DNMT1 and MORC2 causes massive accumulation of L1 transcripts. We identify the same mechanistic hierarchy at pericentromeric α-satellites and clustered protocadherin genes, repetitive elements important for chromosome structure and neurodevelopment respectively. Our data delineate the epigenetic control of repeats in somatic cells, with implications for understanding the vital functions of HUSH-MORC2 in hypomethylated contexts throughout human development.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Sanchez-Luque, F. J. et al. LINE-1 evasion of epigenetic repression in humans. Mol. Cell10.1016/j.molcel.2019.05.024 (2019). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
