

Time-series sewage metagenomics distinguishes seasonal, human-derived and environmental microbial communities potentially allowing source-attributed surveillance
- PMID: 39215001
- PMCID: PMC11364805
- DOI: 10.1038/s41467-024-51957-8
Time-series sewage metagenomics distinguishes seasonal, human-derived and environmental microbial communities potentially allowing source-attributed surveillance
Erratum in
-
Author Correction: Time-series sewage metagenomics distinguishes seasonal, human-derived and environmental microbial communities potentially allowing source-attributed surveillance.Nat Commun. 2024 Oct 17;15(1):8953. doi: 10.1038/s41467-024-53282-6. Nat Commun. 2024. PMID: 39419961 Free PMC article. No abstract available.
Abstract
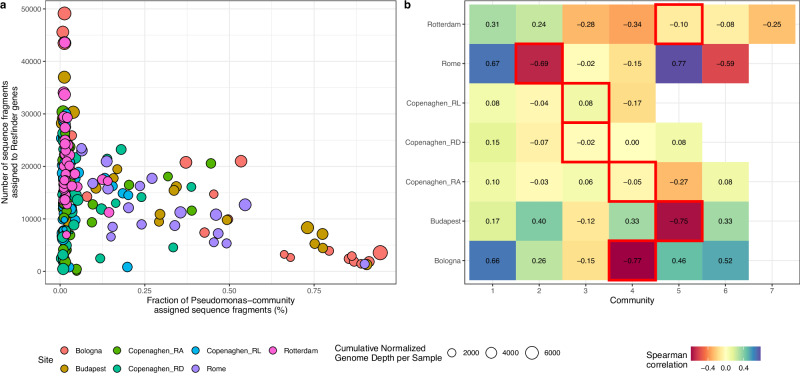
Sewage metagenomics has risen to prominence in urban population surveillance of pathogens and antimicrobial resistance (AMR). Unknown species with similarity to known genomes cause database bias in reference-based metagenomics. To improve surveillance, we seek to recover sewage genomes and develop a quantification and correlation workflow for these genomes and AMR over time. We use longitudinal sewage sampling in seven treatment plants from five major European cities to explore the utility of catch-all sequencing of these population-level samples. Using metagenomic assembly methods, we recover 2332 metagenome-assembled genomes (MAGs) from prokaryotic species, 1334 of which were previously undescribed. These genomes account for ~69% of sequenced DNA and provide insight into sewage microbial dynamics. Rotterdam (Netherlands) and Copenhagen (Denmark) show strong seasonal microbial community shifts, while Bologna, Rome, (Italy) and Budapest (Hungary) have occasional blooms of Pseudomonas-dominated communities, accounting for up to ~95% of sample DNA. Seasonal shifts and blooms present challenges for effective sewage surveillance. We find that bacteria of known shared origin, like human gut microbiota, form communities, suggesting the potential for source-attributing novel species and their ARGs through network community analysis. This could significantly improve AMR tracking in urban environments.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Thomas, K. V. et al. Comparing illicit drug use in 19 European cities through sewage analysis. Sci. Total Environ.432, 432–439 (2012). - PubMed
MeSH terms
Substances
Grants and funding
- NNF16OC0021856/Novo Nordisk Fonden (Novo Nordisk Foundation)
- NNF16OC0021856/Novo Nordisk Fonden (Novo Nordisk Foundation)
- 874735/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- 874735/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
LinkOut - more resources
Full Text Sources
