

An isoform-resolution transcriptomic atlas of colorectal cancer from long-read single-cell sequencing
- PMID: 39216476
- PMCID: PMC11480860
- DOI: 10.1016/j.xgen.2024.100641
An isoform-resolution transcriptomic atlas of colorectal cancer from long-read single-cell sequencing
Abstract
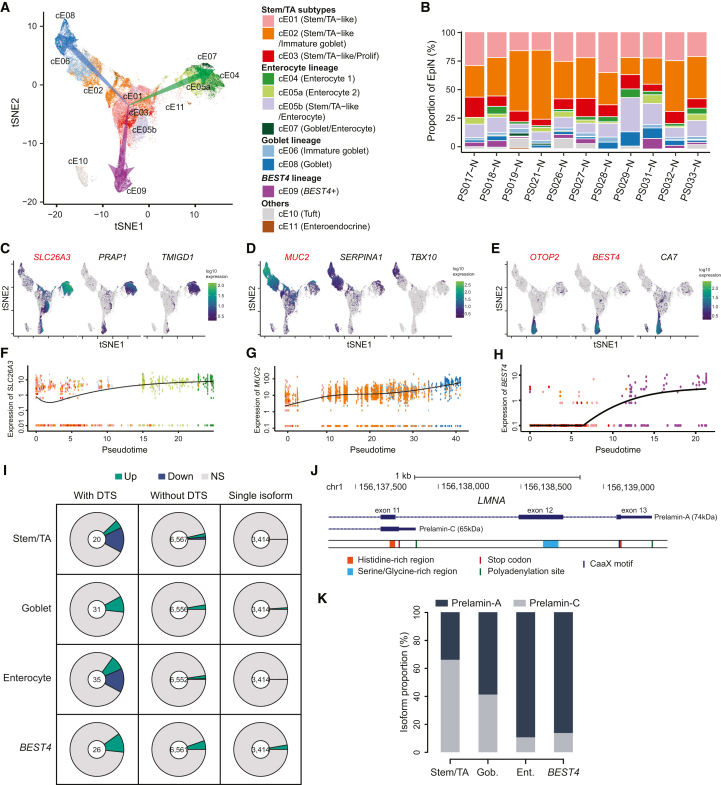
Colorectal cancer (CRC) ranks as the second leading cause of cancer deaths globally. In recent years, short-read single-cell RNA sequencing (scRNA-seq) has been instrumental in deciphering tumor heterogeneities. However, these studies only enable gene-level quantification but neglect alterations in transcript structures arising from alternative end processing or splicing. In this study, we integrated short- and long-read scRNA-seq of CRC samples to build an isoform-resolution CRC transcriptomic atlas. We identified 394 dysregulated transcript structures in tumor epithelial cells, including 299 resulting from various combinations of splicing events. Second, we characterized genes and isoforms associated with epithelial lineages and subpopulations exhibiting distinct prognoses. Among 31,935 isoforms with novel junctions, 330 were supported by The Cancer Genome Atlas RNA-seq and mass spectrometry data. Finally, we built an algorithm that integrated novel peptides derived from open reading frames of recurrent tumor-specific transcripts with mass spectrometry data and identified recurring neoepitopes that may aid the development of cancer vaccines.
Keywords: colorectal cancer; long-read RNA-seq; neoantigen; scRNA-seq.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures







References
-
- Becker W.R., Nevins S.A., Chen D.C., Chiu R., Horning A.M., Guha T.K., Laquindanum R., Mills M., Chaib H., Ladabaum U., et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat. Genet. 2022;54:985–995. doi: 10.1038/s41588-022-01088-x. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
