

A common druggable signature of oncogenic c-Myc, mutant KRAS and mutant p53 reveals functional redundancy and competition among oncogenes in cancer
- PMID: 39217152
- PMCID: PMC11365971
- DOI: 10.1038/s41419-024-06965-3
A common druggable signature of oncogenic c-Myc, mutant KRAS and mutant p53 reveals functional redundancy and competition among oncogenes in cancer
Abstract
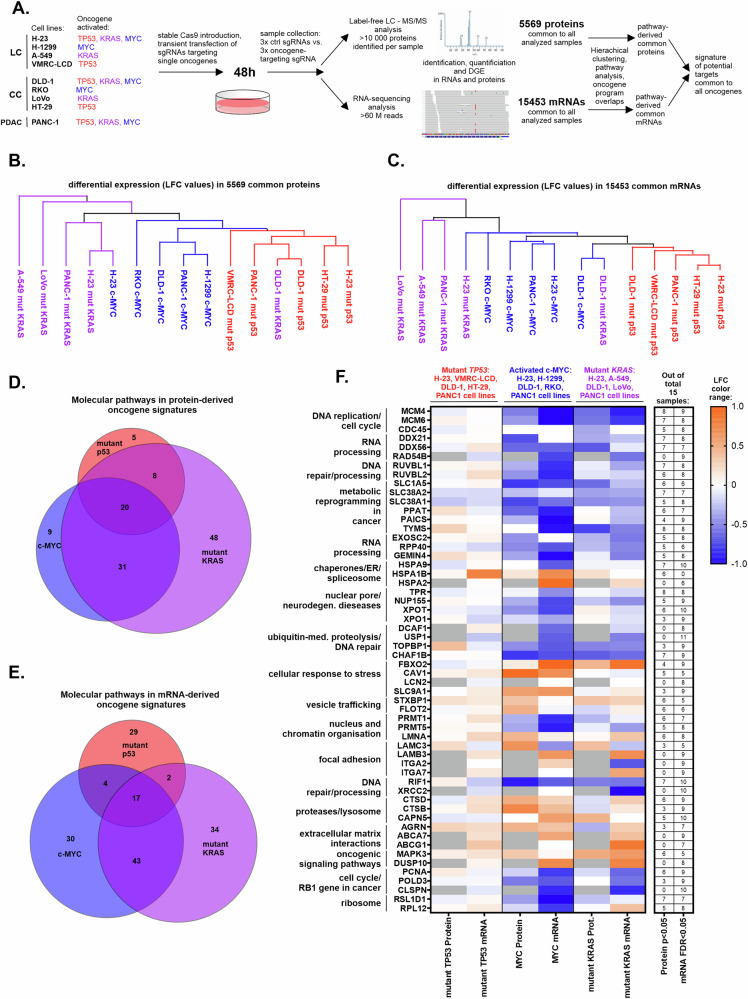
The major driver oncogenes MYC, mutant KRAS, and mutant TP53 often coexist and cooperate to promote human neoplasia, which results in anticancer therapeutic opportunities within their downstream molecular programs. However, little research has been conducted on whether redundancy and competition among oncogenes affect their programs and ability to drive neoplasia. By CRISPR‒Cas9-mediated downregulation we evaluated the downstream proteomics and transcriptomics programs of MYC, mutant KRAS, and mutant TP53 in a panel of cell lines with either one or three of these oncogenes activated, in cancers of the lung, colon and pancreas. Using RNAi screening of the commonly activated molecular programs, we found a signature of three proteins - RUVBL1, HSPA9, and XPO1, which could be efficiently targeted by novel drug combinations in the studied cancer types. Interestingly, the signature was controlled by the oncoproteins in a redundant or competitive manner rather than by cooperation. Each oncoprotein individually upregulated the target genes, while upon oncogene co-expression each target was controlled preferably by a dominant oncoprotein which reduced the influence of the others. This interplay was mediated by redundant routes of target gene activation - as in the case of mutant KRAS signaling to c-Jun/GLI2 transcription factors bypassing c-Myc activation, and by competition - as in the case of mutant p53 and c-Myc competing for binding to target promoters. The global transcriptomics data from the cell lines and patient samples indicate that the redundancy and competition of oncogenic programs are broad phenomena, that may constitute even a majority of the genes dependent on oncoproteins, as shown for mutant p53 in colon and lung cancer cell lines. Nevertheless, we demonstrated that redundant oncogene programs harbor targets for efficient anticancer drug combinations, bypassing the limitations for direct oncoprotein inhibition.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
MeSH terms
Substances
Grants and funding
- 2017/26/E/NZ5/00663/Narodowe Centrum Nauki (National Science Centre)
- PTO/Servier oncology grant ed.2022/23/Servier (Laboratoires Servier)
- FBW-03/2021/Instytut Medycyny Doswiadczalnej i Klinicznej im. M. Mossakowskiego, Polskiej Akademii Nauk (Mossakowski Medical Research Centre, Polish Academy of Sciences)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
