

scEpiAge: an age predictor highlighting single-cell ageing heterogeneity in mouse blood
- PMID: 39217176
- PMCID: PMC11366017
- DOI: 10.1038/s41467-024-51833-5
scEpiAge: an age predictor highlighting single-cell ageing heterogeneity in mouse blood
Abstract
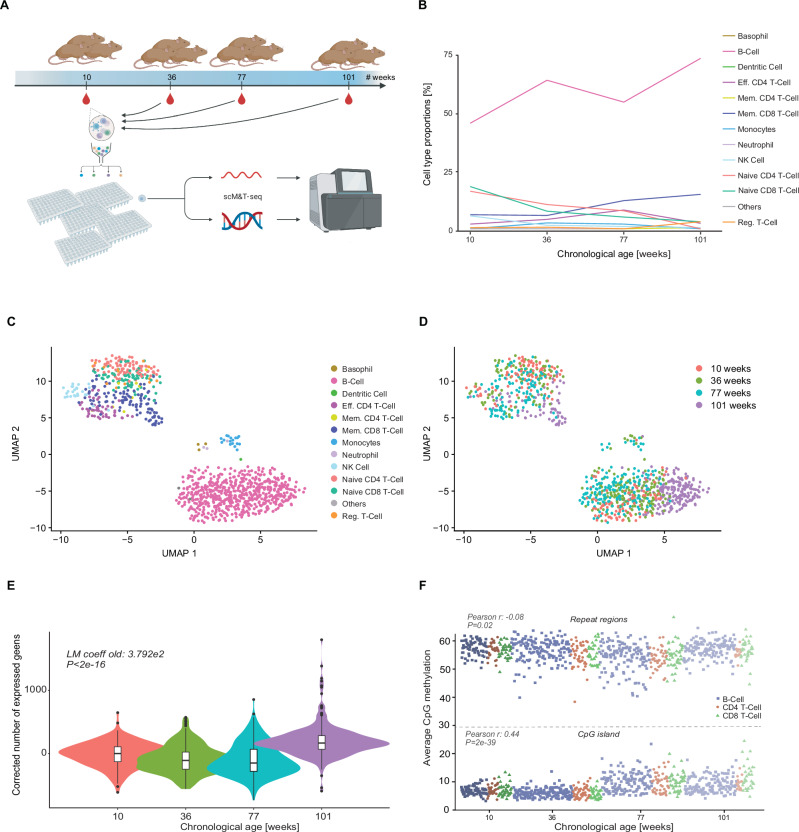
Ageing is the accumulation of changes and decline of function of organisms over time. The concept and biomarkers of biological age have been established, notably DNA methylation-based clocks. The emergence of single-cell DNA methylation profiling methods opens the possibility of studying the biological age of individual cells. Here, we generate a large single-cell DNA methylation and transcriptome dataset from mouse peripheral blood samples, spanning a broad range of ages. The number of genes expressed increases with age, but gene-specific changes are small. We next develop scEpiAge, a single-cell DNA methylation age predictor, which can accurately predict age in (very sparse) publicly available datasets, and also in single cells. DNA methylation age distribution is wider than technically expected, indicating epigenetic age heterogeneity and functional differences. Our work provides a foundation for single-cell and sparse data epigenetic age predictors, validates their functionality and highlights epigenetic heterogeneity during ageing.
© 2024. The Author(s).
Conflict of interest statement
W.R. is a consultant and shareholder of Cambridge Epigenetix. S.C., F.K., and W.R. are employees of Altos Labs. T.S. is founder and CEO of Chronomics Ltd. O.S. is a paid consultant of Insitro.INC. F.v.M. is a consultant and shareholder of Longevity Consultancy Group Sàrl. The remaining authors declare no competing interests.
Figures




References
MeSH terms
Grants and funding
- BBS/E/B/000C0422/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- BBS/E/B/000C0421/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- 803491/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- WT_/Wellcome Trust/United Kingdom
- 950349/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
LinkOut - more resources
Full Text Sources
Medical
