

Mitophagy and cGAS-STING crosstalk in neuroinflammation
- PMID: 39220869
- PMCID: PMC11365416
- DOI: 10.1016/j.apsb.2024.05.012
Mitophagy and cGAS-STING crosstalk in neuroinflammation
Abstract
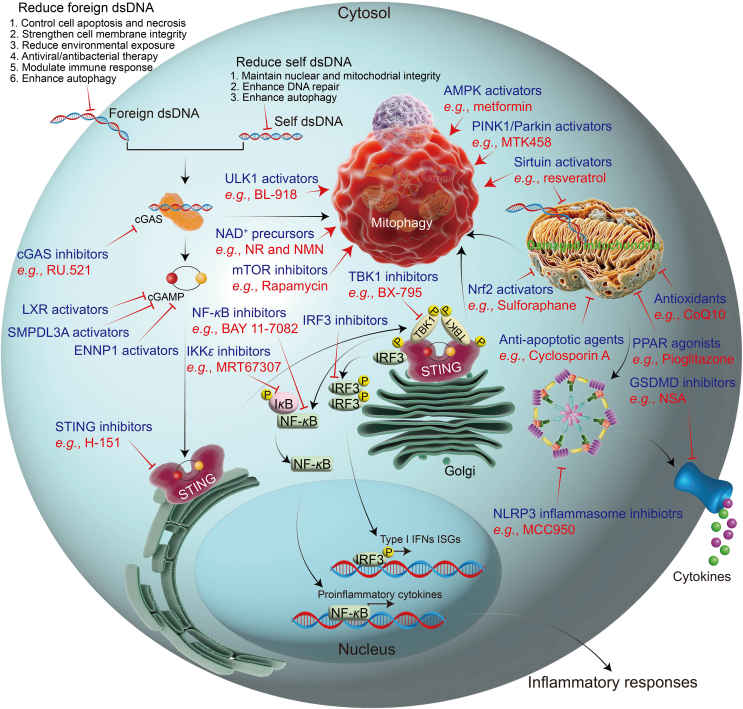
Mitophagy, essential for mitochondrial health, selectively degrades damaged mitochondria. It is intricately linked to the cGAS-STING pathway, which is crucial for innate immunity. This pathway responds to mitochondrial DNA and is associated with cellular stress response. Our review explores the molecular details and regulatory mechanisms of mitophagy and the cGAS-STING pathway. We critically evaluate the literature demonstrating how dysfunctional mitophagy leads to neuroinflammatory conditions, primarily through the accumulation of damaged mitochondria, which activates the cGAS-STING pathway. This activation prompts the production of pro-inflammatory cytokines, exacerbating neuroinflammation. This review emphasizes the interaction between mitophagy and the cGAS-STING pathways. Effective mitophagy may suppress the cGAS-STING pathway, offering protection against neuroinflammation. Conversely, impaired mitophagy may activate the cGAS-STING pathway, leading to chronic neuroinflammation. Additionally, we explored how this interaction influences neurodegenerative disorders, suggesting a common mechanism underlying these diseases. In conclusion, there is a need for additional targeted research to unravel the complexities of mitophagy-cGAS-STING interactions and their role in neurodegeneration. This review highlights potential therapies targeting these pathways, potentially leading to new treatments for neuroinflammatory and neurodegenerative conditions. This synthesis enhances our understanding of the cellular and molecular foundations of neuroinflammation and opens new therapeutic avenues for neurodegenerative disease research.
Keywords: Crosstalk; Innate immunity; Mitochondrial DNA; Mitophagy; Neurodegenerative diseases; Neuroinflammation; Therapeutic avenues; cGAS–STING.
© 2024 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures








References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
