

Understanding and Overcoming Resistance to Selective FGFR Inhibitors across FGFR2-Driven Malignancies
- PMID: 39226398
- PMCID: PMC7616615
- DOI: 10.1158/1078-0432.CCR-24-1834
Understanding and Overcoming Resistance to Selective FGFR Inhibitors across FGFR2-Driven Malignancies
Abstract
Purpose: Understanding resistance to selective FGFR inhibitors is crucial to improve the clinical outcomes of patients with FGFR2-driven malignancies.
Experimental design: We analyzed sequential ctDNA, ± whole-exome sequencing, or targeted next-generation sequencing on tissue biopsies from patients with tumors harboring activating FGFR2 alterations progressing on pan-FGFR-selective inhibitors, collected in the prospective UNLOCK program. FGFR2::BICC1 Ba/F3 and patient-derived xenograft models were used for functional studies.
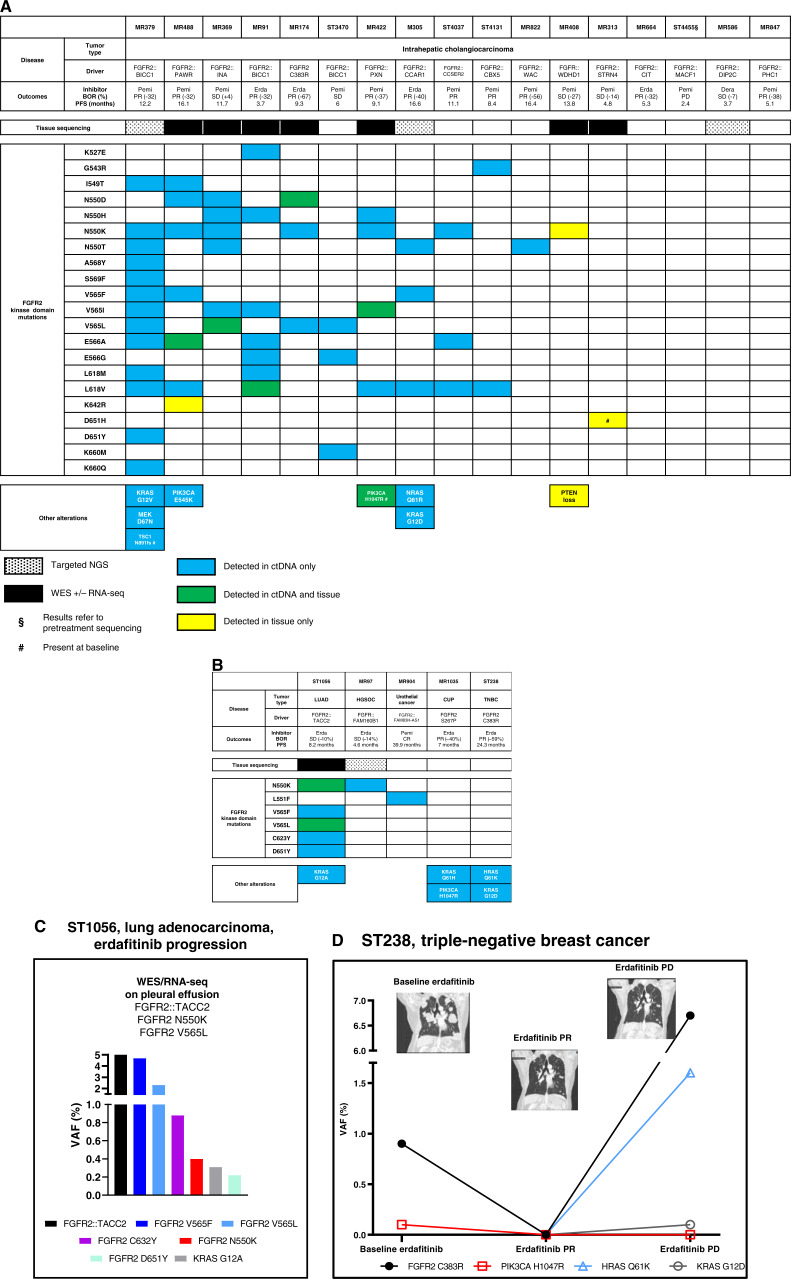
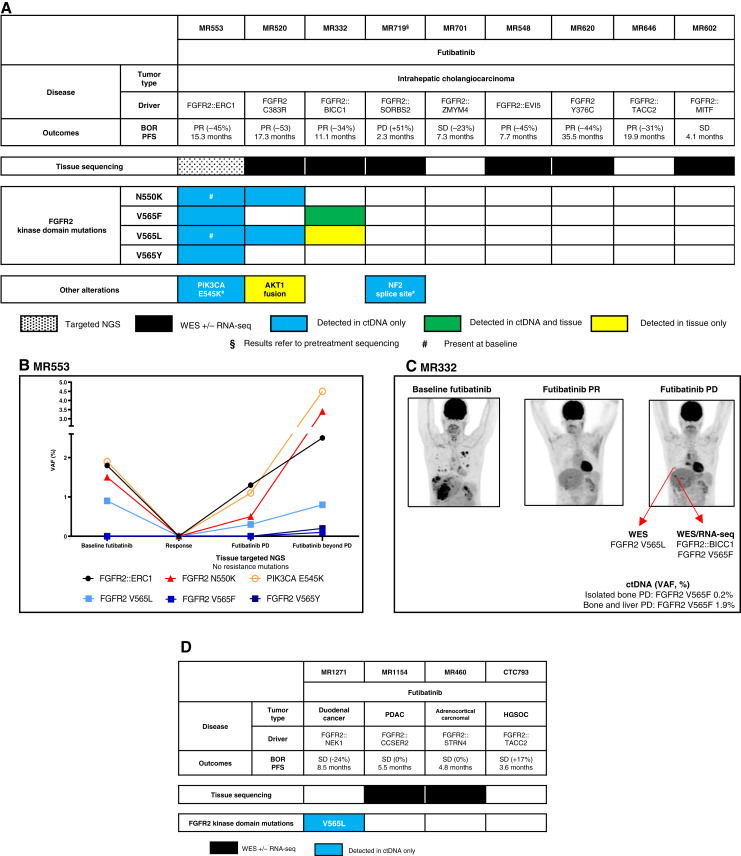
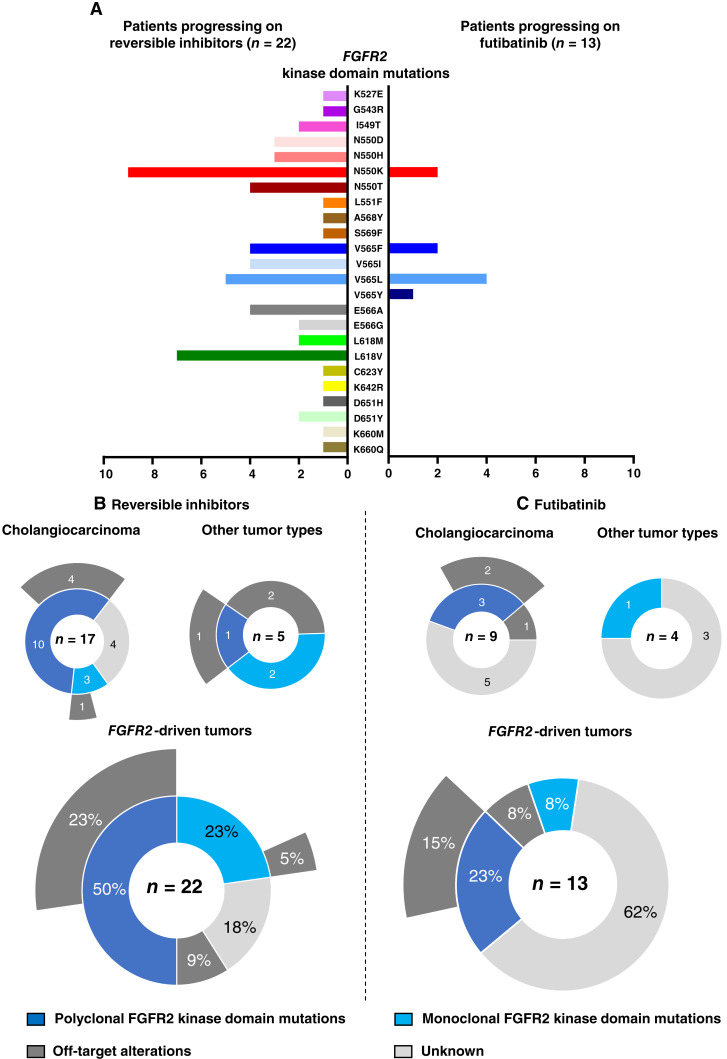
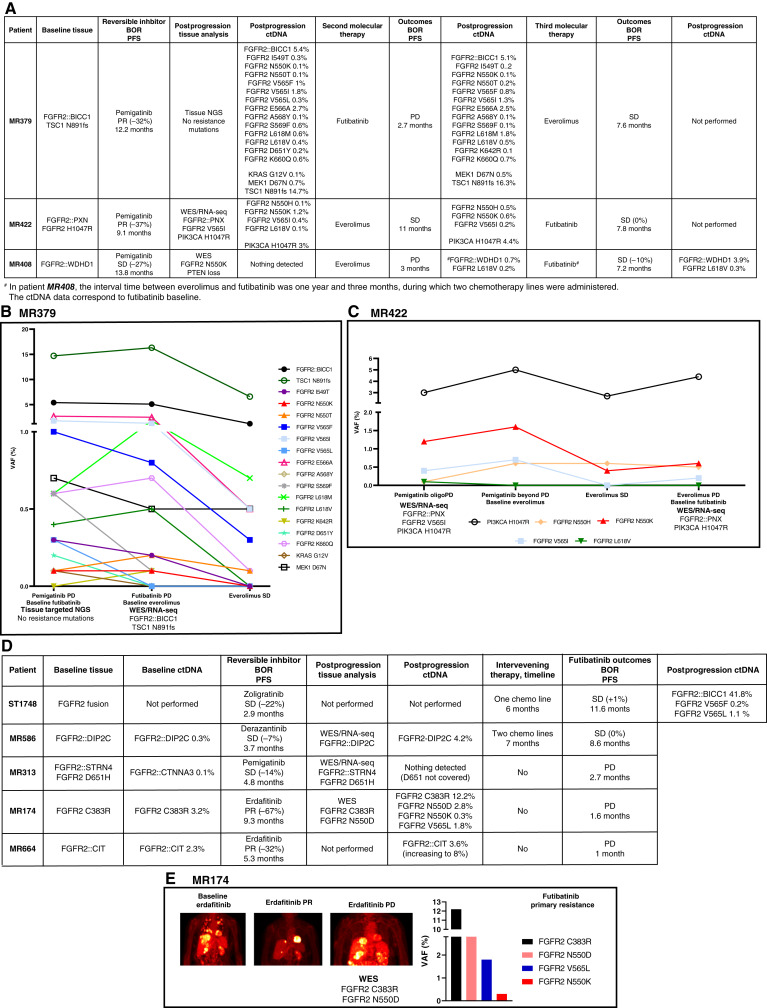
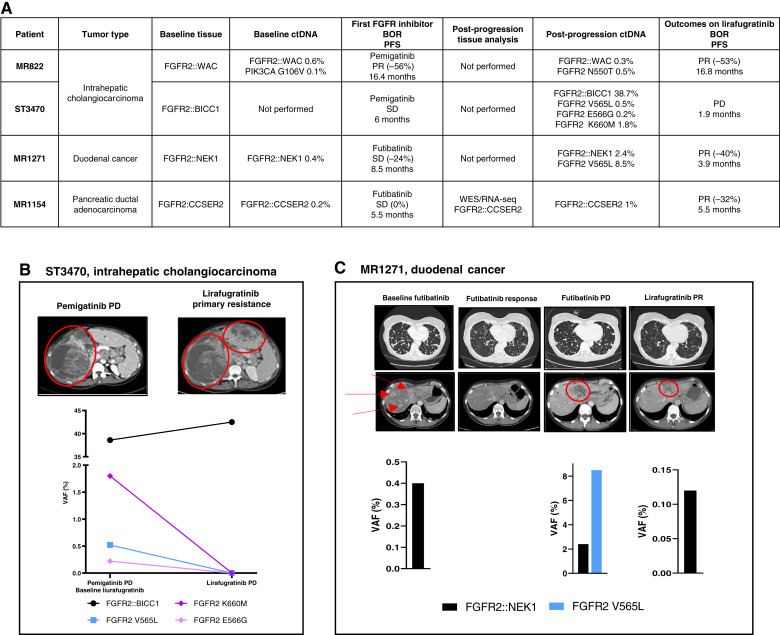
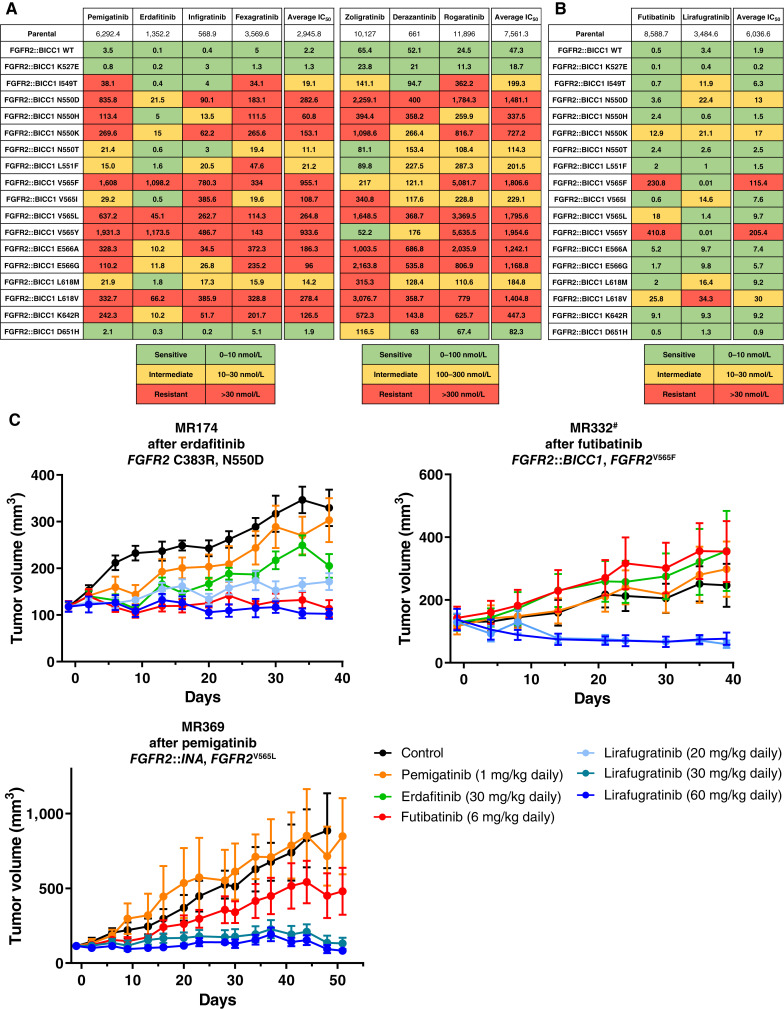
Results: Thirty-six patients were included. In cholangiocarcinoma, at resistance to both reversible inhibitors (e.g., pemigatinib and erdafitinib) and the irreversible inhibitor futibatinib, polyclonal FGFR2 kinase domain mutations were frequent (14/27 patients). Tumors other than cholangiocarcinoma shared the same mutated FGFR2 residues, but polyclonality was rare (1/9 patients). At resistance to reversible inhibitors, 14 residues in the FGFR2 kinase domain were mutated-after futibatinib, only the molecular brake N550 and the gatekeeper V565. Off-target alterations in PI3K/mTOR and MAPK pathways were found in 11 patients, often together with on-target mutations. At progression to a first FGFR inhibitor, 12 patients received futibatinib or lirafugratinib (irreversible inhibitors), with variable clinical outcomes depending on previous resistance mechanisms. Two patients with TSC1 or PIK3CA mutations benefited from everolimus. In cell viability assays on Ba/F3 and in pharmacologic studies on patient-derived xenografts, irreversible inhibitors retained better activity against FGFR2 kinase domain mutations, with lirafugratinib active against the recalcitrant V565L/F/Y.
Conclusions: At progression to FGFR inhibitors, FGFR2-driven malignancies are characterized by high intra- and interpatient molecular heterogeneity, particularly in cholangiocarcinoma. Resistance to FGFR inhibitors can be overcome by sequential, molecularly oriented treatment strategies across FGFR2-driven tumors.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
F. Facchinetti reports personal fees from BeiGene outside the submitted work. Y. Loriot reports personal fees, nonfinancial support, and other support from Janssen during the conduct of the study, as well as personal fees, nonfinancial support, and other support from MSD, Pfizer, Merck KGaA, Astellas, Gilead, Bristol Myers Squibb, and Roche, nonfinancial support and other support from Incyte, other support from Exelixis, and personal fees and other support from Taiho outside the submitted work. S. Michiels reports personal fees from DSMB or scientific committee study membership for Kedrion, Biophytis, Servier, IQVIA, Yuhan, and Roche outside the submitted work. A. Italiano reports grants and personal fees from Bayer, IPSEN, Roche, and Bristol Myers Squibb and grants from MSD, Merck, AstraZeneca, and BeiGene outside the submitted work. C. Smolenschi reports personal fees from Servier and Merck, nonfinancial support from Astellas and Bristol Myers Squibb, and grants from Pierre Fabre outside the submitted work. L. Tselikas reports personal fees from Incyte during the conduct of the study, as well as personal fees from GE Healthcare, Quantum Surgical, and Boston Scientific outside the submitted work. B. Besse reports other support from AbbVie, BioNTech SE, Bristol Myers Squibb, Chugai Pharmaceutical, CureVac AG, Daiichi Sankyo, F. Hoffmann-La Roche Ltd, PharmaMar, Regeneron, Sanofi-Aventis, Turning Point Therapeutics, Eli Lilly and Company, Ellipses Pharma Ltd, Genmab, Immunocore, Janssen, MSD, Ose Immunotherapeutics, Owkin, Taiho Oncology, AstraZeneca, BeiGene, GlaxoSmithKline, Roche-Genentech, Takeda, Hedera Dx, and Springer Healthcare Ltd during the conduct of the study. F. André reports grants from AstraZeneca, Guardant Health, Novartis, Owkin, Pfizer, Eli Lilly and Company, Roche, and Daiichi Sankyo, other support from Boston Pharmaceutics, Gilead, and Servier, and grants and other support from N-Power Medicine outside the submitted work. A. Hollebecque reports personal fees and nonfinancial support from Amgen, personal fees from Basilea, Bristol Myers Squibb, Servier, Relay Therapeutics, Taiho, MSD, Seagen, grants and personal fees from Incyte, and nonfinancial support from Pierre Fabre outside the submitted work, as well as reports being a PI of the TransThera (Tinengotinib) phase III trial. L. Friboulet reports grants from Relay Therapeutics during the conduct of the study, as well as grants from Nuvalent and Sanofi outside the submitted work. No disclosures were reported by the other authors.
Figures
References
-
- Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259–67. - PubMed
-
- Graham RP, Barr Fritcher EG, Pestova E, Schulz J, Sitailo LA, Vasmatzis G, et al. . Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630–8. - PubMed
-
- Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. . Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427–34. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
