

A framework for sharing of clinical and genetic data for precision medicine applications
- PMID: 39227443
- PMCID: PMC11645287
- DOI: 10.1038/s41591-024-03239-5
A framework for sharing of clinical and genetic data for precision medicine applications
Abstract
Precision medicine has the potential to provide more accurate diagnosis, appropriate treatment and timely prevention strategies by considering patients' biological makeup. However, this cannot be realized without integrating clinical and omics data in a data-sharing framework that achieves large sample sizes. Systems that integrate clinical and genetic data from multiple sources are scarce due to their distinct data types, interoperability, security and data ownership issues. Here we present a secure framework that allows immutable storage, querying and analysis of clinical and genetic data using blockchain technology. Our platform allows clinical and genetic data to be harmonized by combining them under a unified framework. It supports combined genotype-phenotype queries and analysis, gives institutions control of their data and provides immutable user access logs, improving transparency into how and when health information is used. We demonstrate the value of our framework for precision medicine by creating genotype-phenotype cohorts and examining relationships within them. We show that combining data across institutions using our secure platform increases statistical power for rare disease analysis. By offering an integrated, secure and decentralized framework, we aim to enhance reproducibility and encourage broader participation from communities and patients in data sharing.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: A patent application has been filed by Columbia University with A.E. and G.G listed as inventors (application number: 18/419,923; status of application: pending; specific aspect of manuscript covered in patent application: blockchain-based harmonization of clinical and genetic data). All other authors declare no competing interests.
Figures
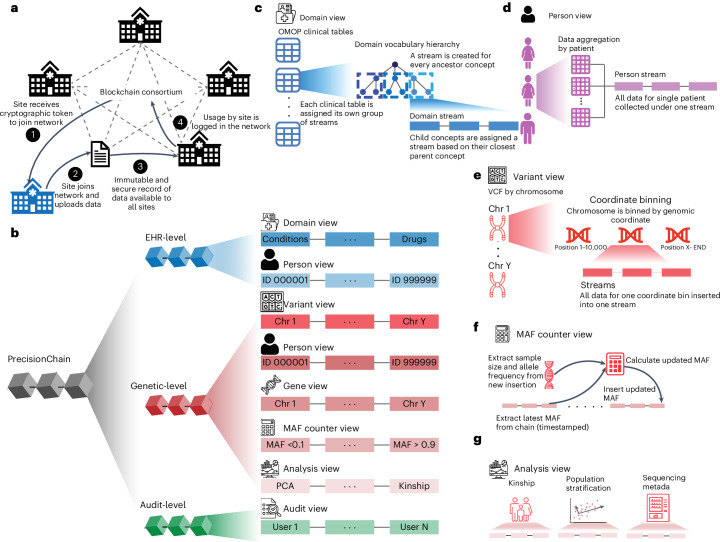
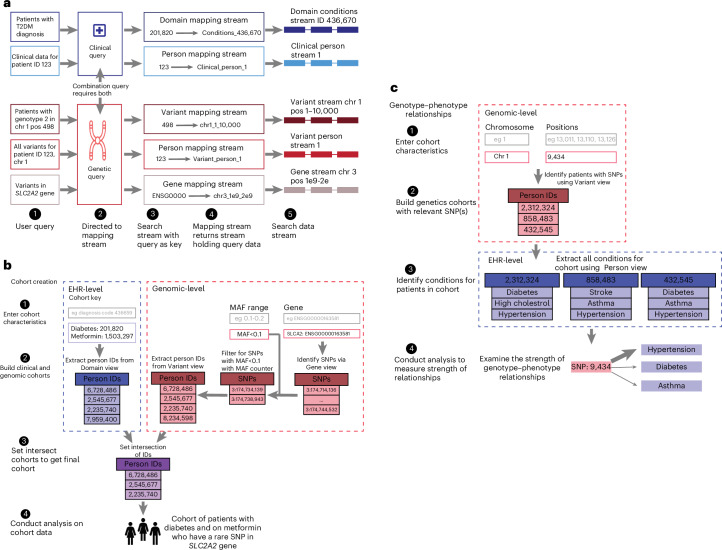
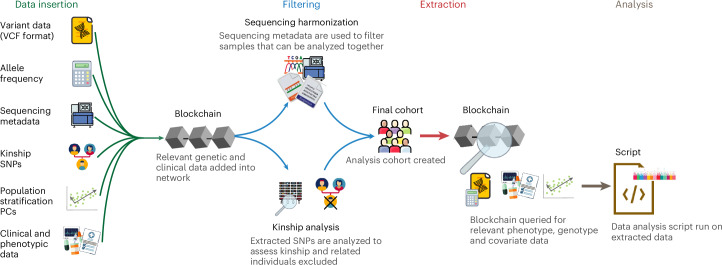
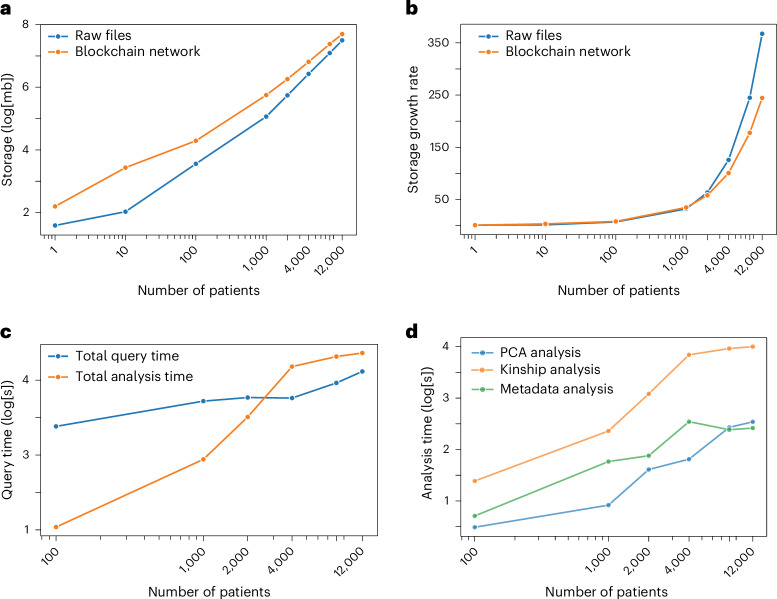
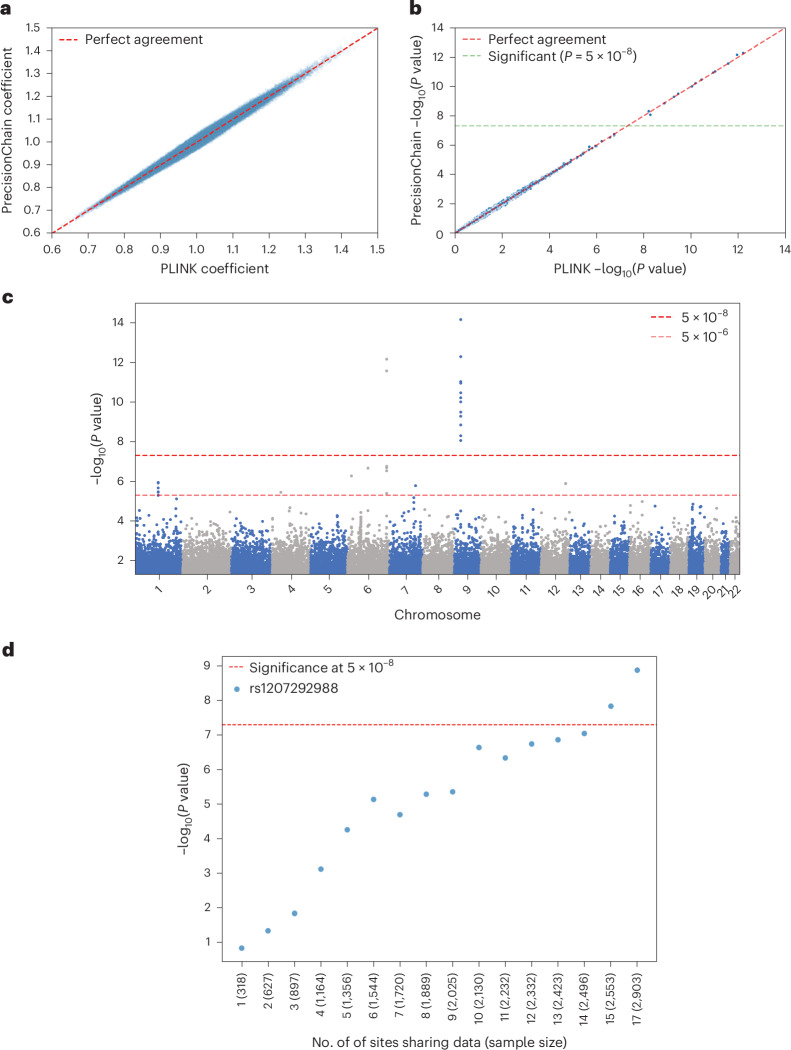
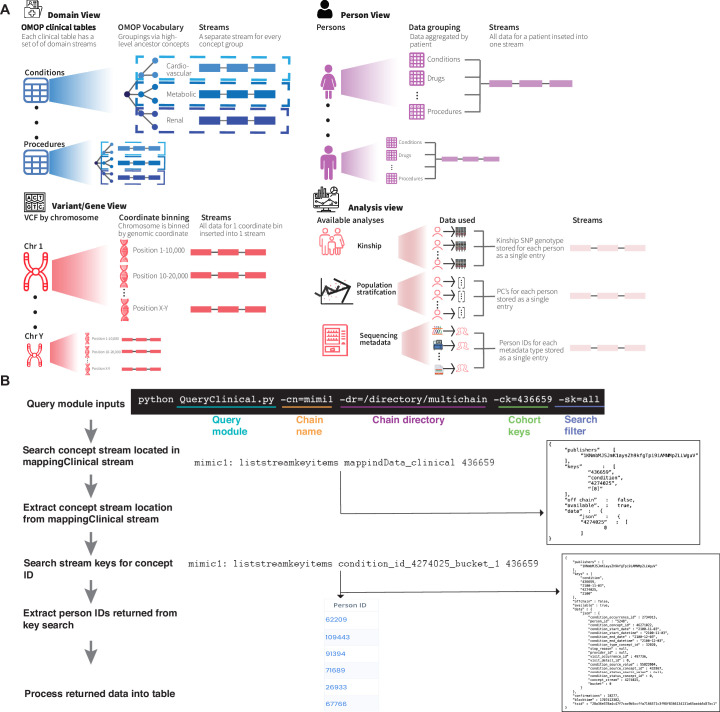
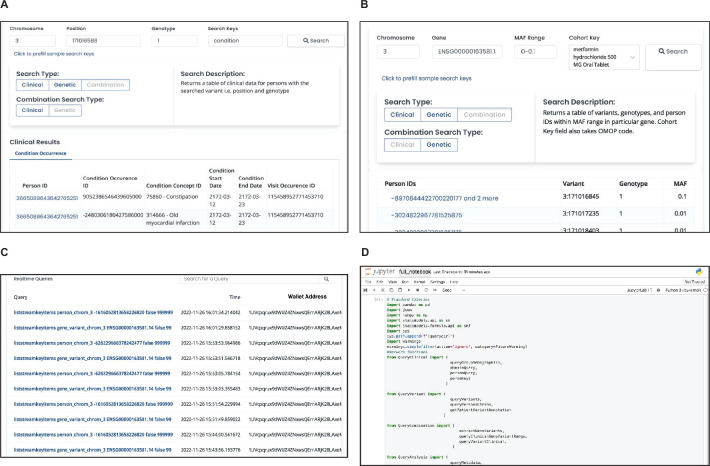
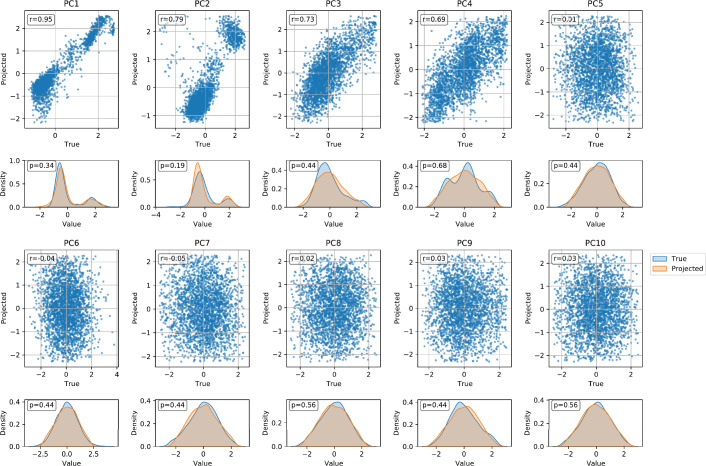
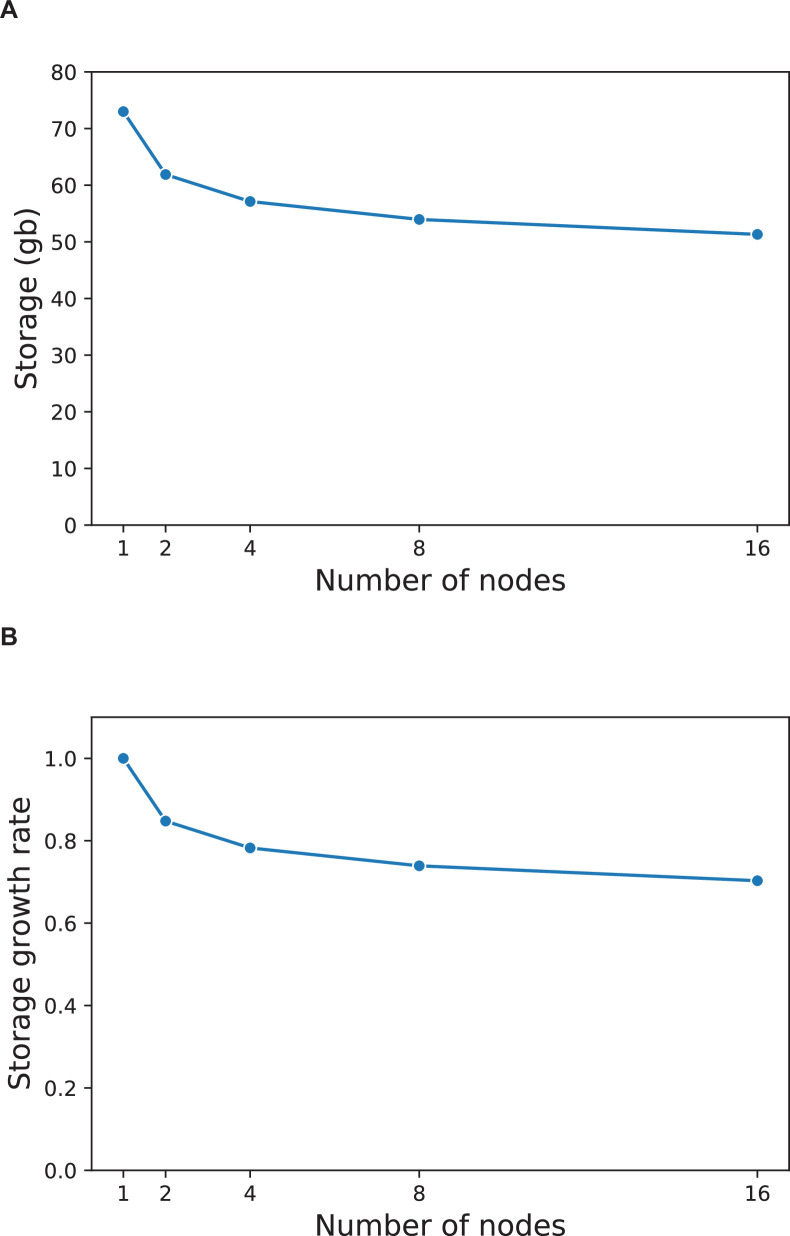
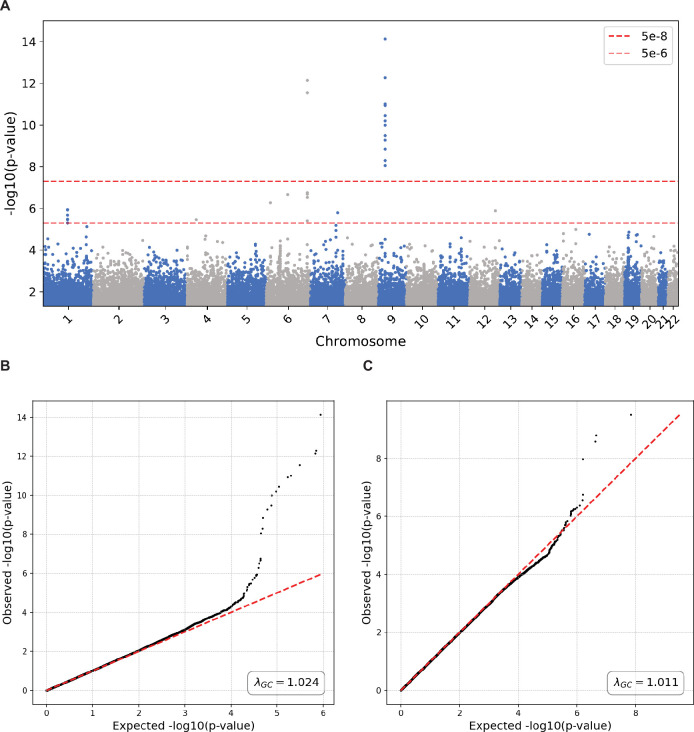
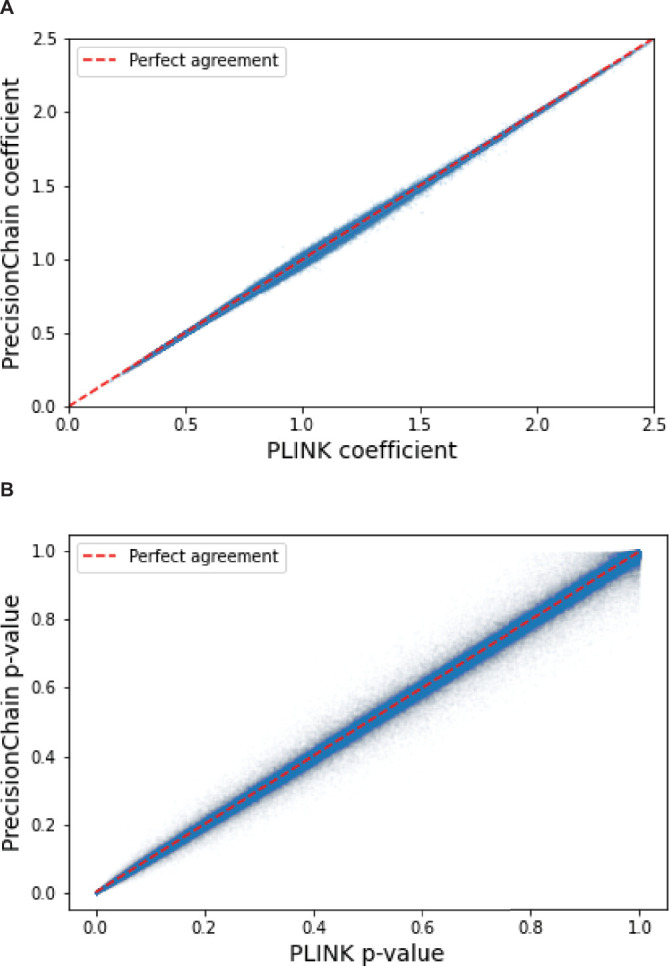
References
-
- Ward, R. & Ginsburg, G. S. Local and global challenges in the clinical implementation of precision medicine. In Genomic and Precision Medicine: Foundations, Translation, and Implementation 3rd edn (eds Ginsburg G. S. & Willard, H. F.) 105–117 (Academic Press, 2016).
-
- Precision Cancer Medicine: Challenges and Opportunities (eds Roychowdhury, S. & Van Allen, E. M.) (Springer, 2020).
-
- Acosta, J. N., Falcone, G. J., Rajpurkar, P. & Topol, E. J. Multimodal biomedical AI. Nat. Med.28, 1773–1784 (2022). - PubMed
MeSH terms
Grants and funding
- R35 GM147004/GM/NIGMS NIH HHS/United States
- R35GM147004/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R00HG010909/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R00 HG010909/HG/NHGRI NIH HHS/United States
- P30 AG066514/AG/NIA NIH HHS/United States
