

This is a preprint.
The Genetic Determinants and Genomic Consequences of Non-Leukemogenic Somatic Point Mutations
- PMID: 39228737
- PMCID: PMC11370504
- DOI: 10.1101/2024.08.22.24312319
The Genetic Determinants and Genomic Consequences of Non-Leukemogenic Somatic Point Mutations
Update in
-
Genetic determinants and genomic consequences of non-leukemogenic somatic point mutations.Nat Commun. 2025 Oct 16;16(1):9194. doi: 10.1038/s41467-025-64236-x. Nat Commun. 2025. PMID: 41102182 Free PMC article.
Abstract
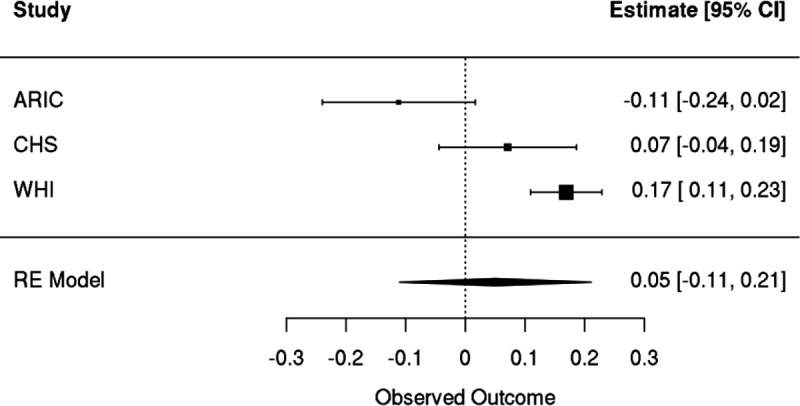
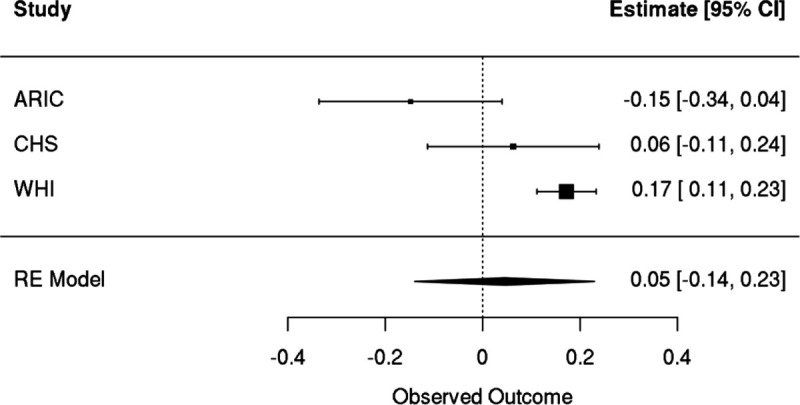
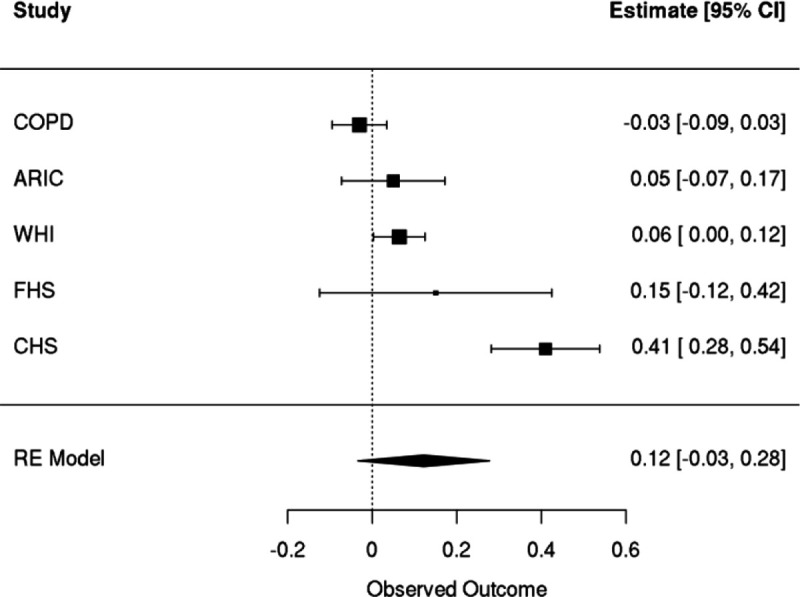
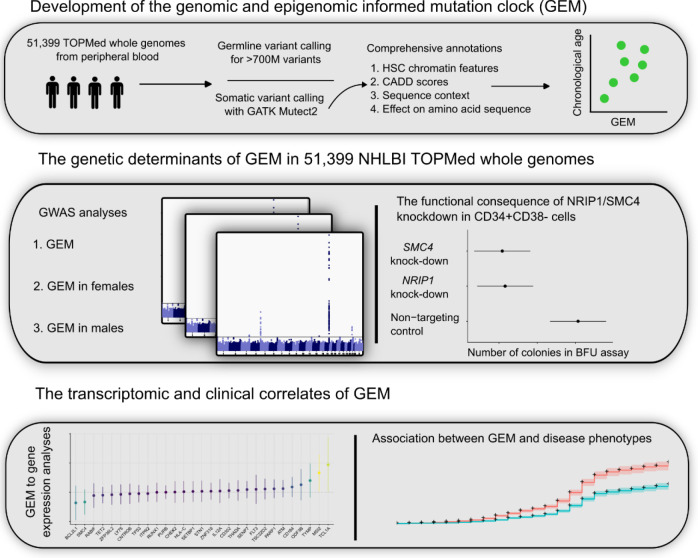
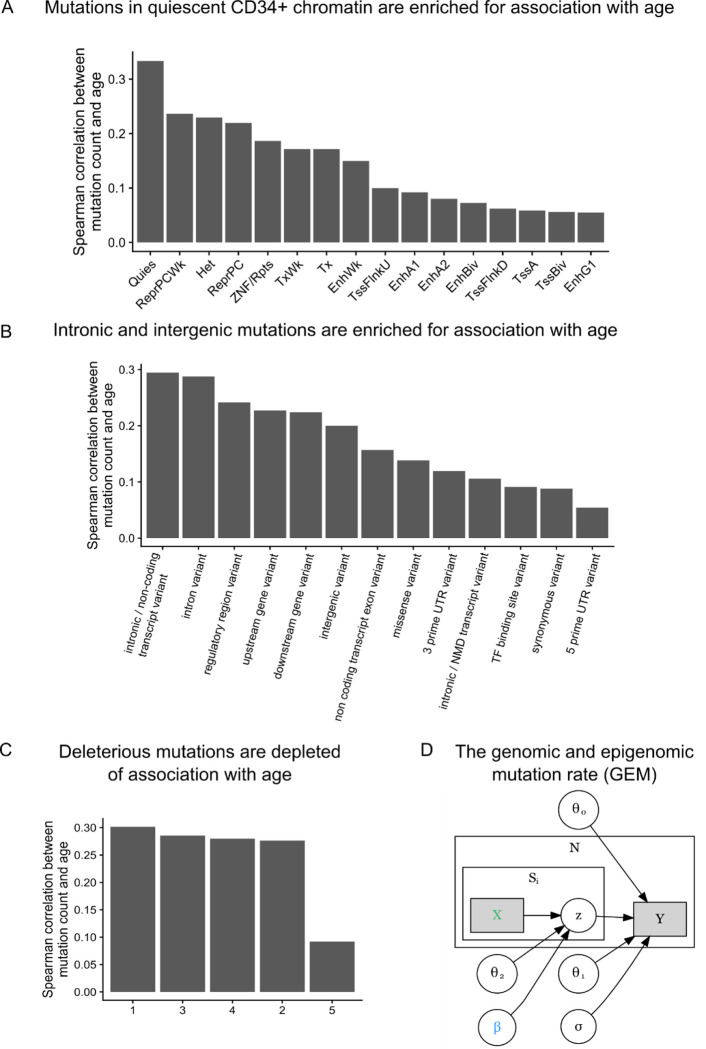
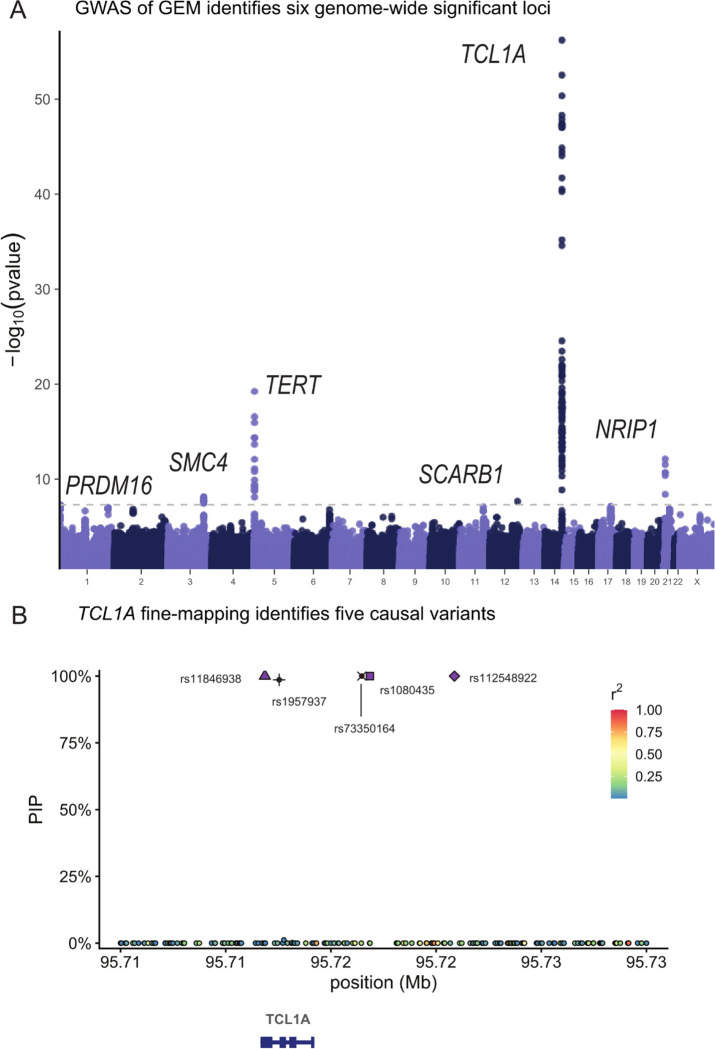
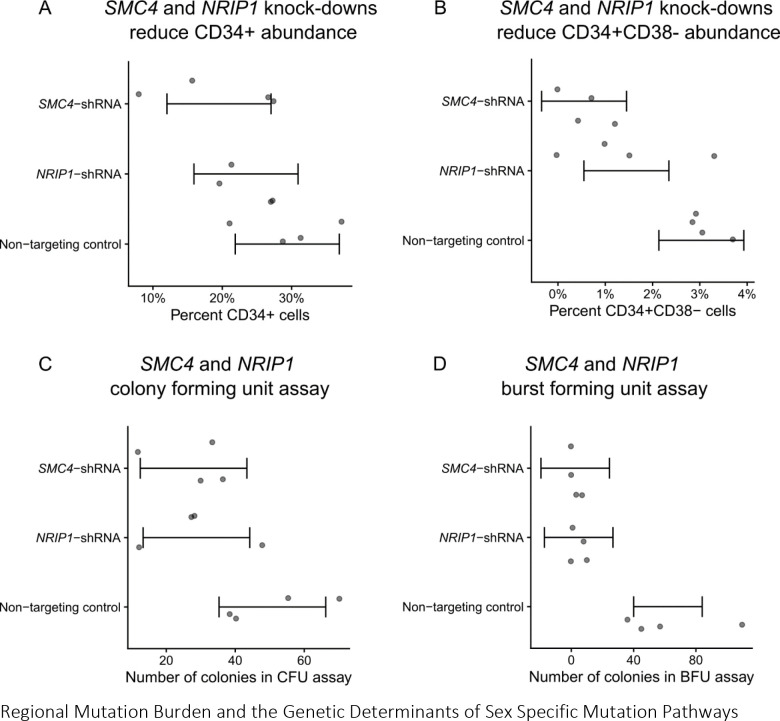
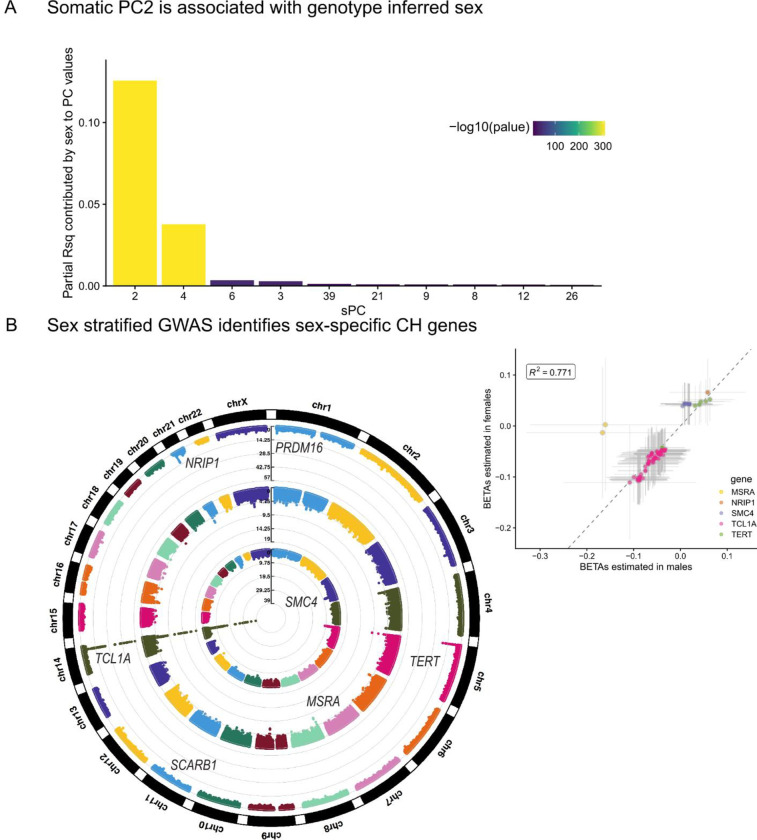
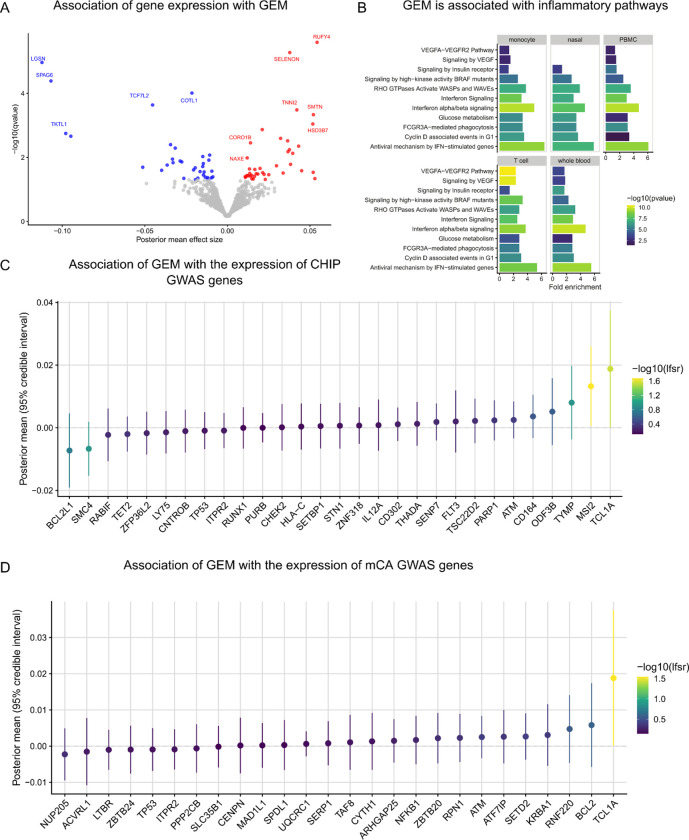
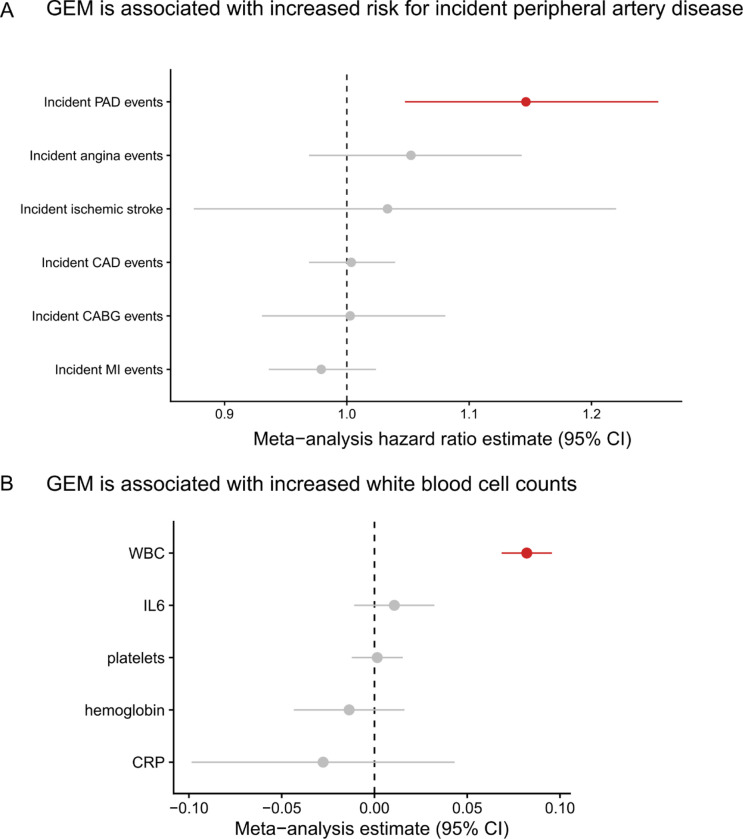
Clonal hematopoiesis (CH) is defined by the expansion of a lineage of genetically identical cells in blood. Genetic lesions that confer a fitness advantage, such as point mutations or mosaic chromosomal alterations (mCAs) in genes associated with hematologic malignancy, are frequent mediators of CH. However, recent analyses of both single cell-derived colonies of hematopoietic cells and population sequencing cohorts have revealed CH frequently occurs in the absence of known driver genetic lesions. To characterize CH without known driver genetic lesions, we used 51,399 deeply sequenced whole genomes from the NHLBI TOPMed sequencing initiative to perform simultaneous germline and somatic mutation analyses among individuals without leukemogenic point mutations (LPM), which we term CH-LPMneg. We quantified CH by estimating the total mutation burden. Because estimating somatic mutation burden without a paired-tissue sample is challenging, we developed a novel statistical method, the Genomic and Epigenomic informed Mutation (GEM) rate, that uses external genomic and epigenomic data sources to distinguish artifactual signals from true somatic mutations. We performed a genome-wide association study of GEM to discover the germline determinants of CH-LPMneg. After fine-mapping and variant-to-gene analyses, we identified seven genes associated with CH-LPMneg (TCL1A, TERT, SMC4, NRIP1, PRDM16, MSRA, SCARB1), and one locus associated with a sex-associated mutation pathway (SRGAP2C). We performed a secondary analysis excluding individuals with mCAs, finding that the genetic architecture was largely unaffected by their inclusion. Functional analyses of SMC4 and NRIP1 implicated altered HSC self-renewal and proliferation as the primary mediator of mutation burden in blood. We then performed comprehensive multi-tissue transcriptomic analyses, finding that the expression levels of 404 genes are associated with GEM. Finally, we performed phenotypic association meta-analyses across four cohorts, finding that GEM is associated with increased white blood cell count and increased risk for incident peripheral artery disease, but is not significantly associated with incident stroke or coronary disease events. Overall, we develop GEM for quantifying mutation burden from WGS without a paired-tissue sample and use GEM to discover the genetic, genomic, and phenotypic correlates of CH-LPMneg.
Conflict of interest statement
Competing Interests Declaration L.M.R. is a consultant for the TOPMed Administrative Coordinating Center (through Westat). B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. J.Y. reports grant support from Bayer. M.C. reports grant support from Bayer and GSK, Consulting and speaking fees from Illumina and AstraZeneca. A.G.B., P.N, and S.J. are cofounders, equity holders, and on the scientific advisory board of TenSixteen Bio. G.R.A. is an employee of Regeneron Pharmaceuticals and receives salary, stock and stock options as compensation.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials