

This is a preprint.
Dysfunctional mechanotransduction regulates the progression of PIK3CA-driven vascular malformations
- PMID: 39229154
- PMCID: PMC11370454
- DOI: 10.1101/2024.08.22.609165
Dysfunctional mechanotransduction regulates the progression of PIK3CA-driven vascular malformations
Update in
-
Dysfunctional mechanotransduction regulates the progression of PIK3CA-driven vascular malformations.APL Bioeng. 2025 Feb 5;9(1):016106. doi: 10.1063/5.0234507. eCollection 2025 Mar. APL Bioeng. 2025. PMID: 39935869 Free PMC article.
Abstract
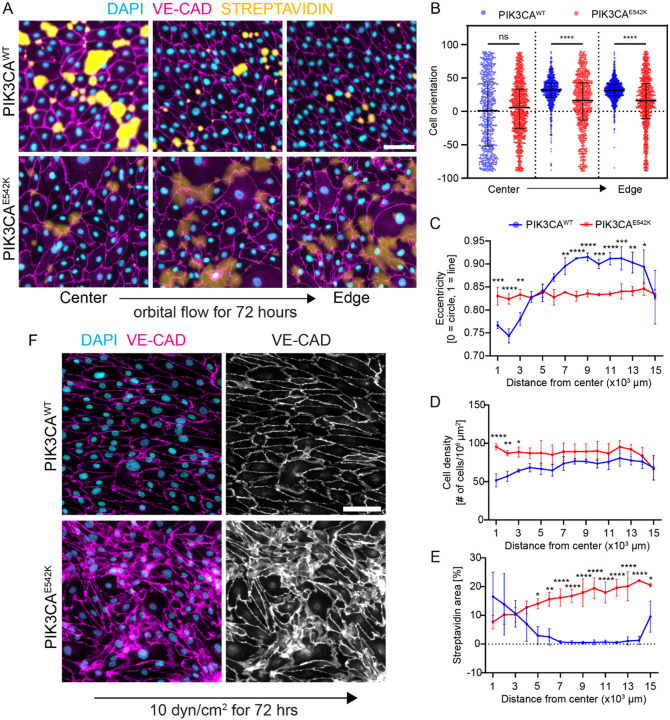
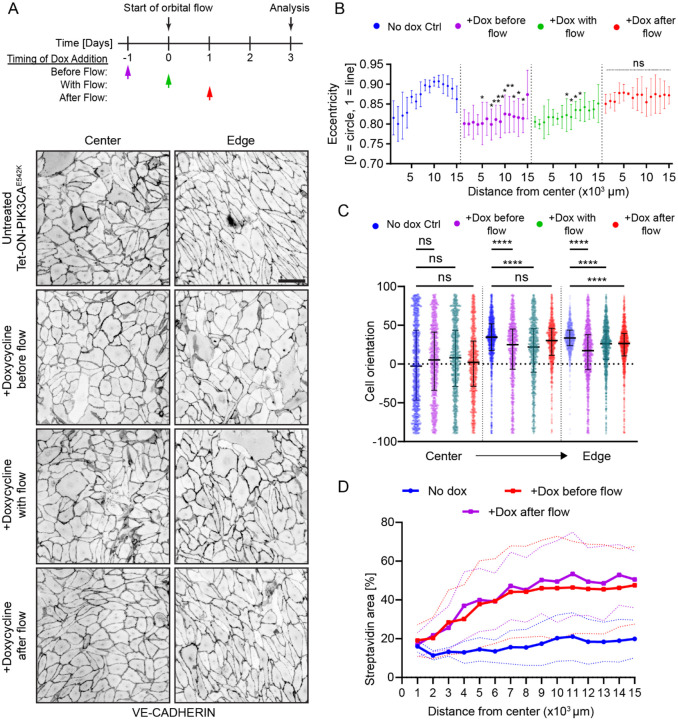
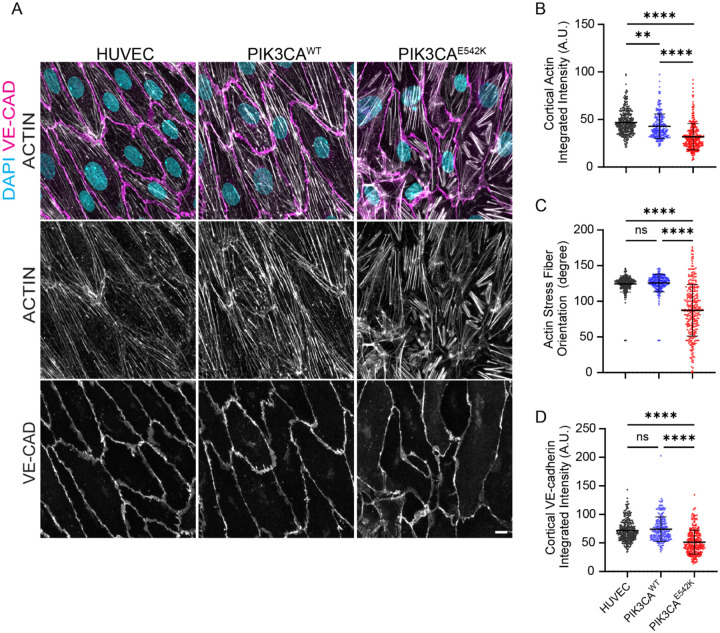
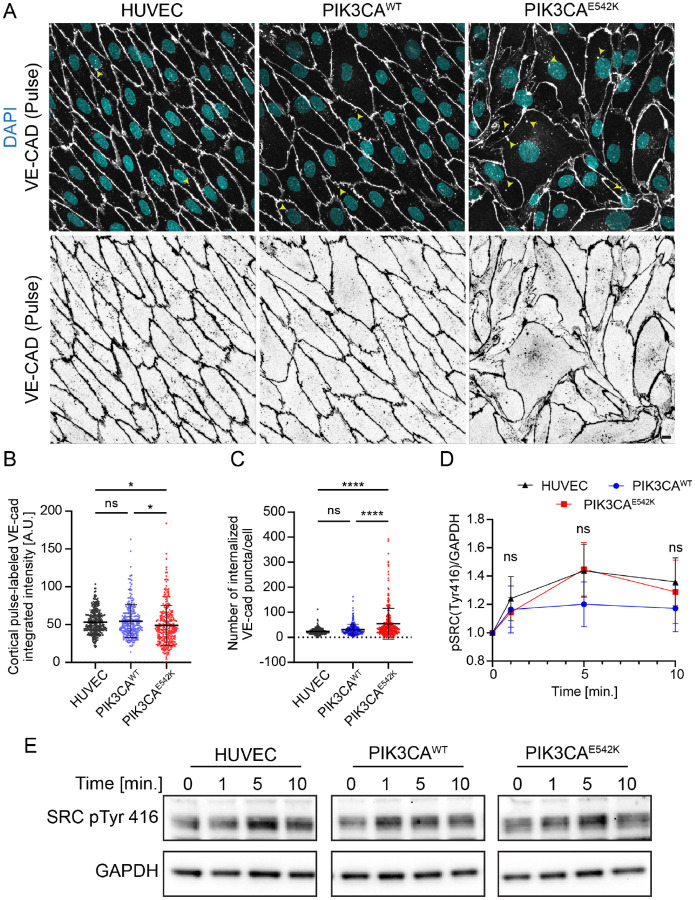
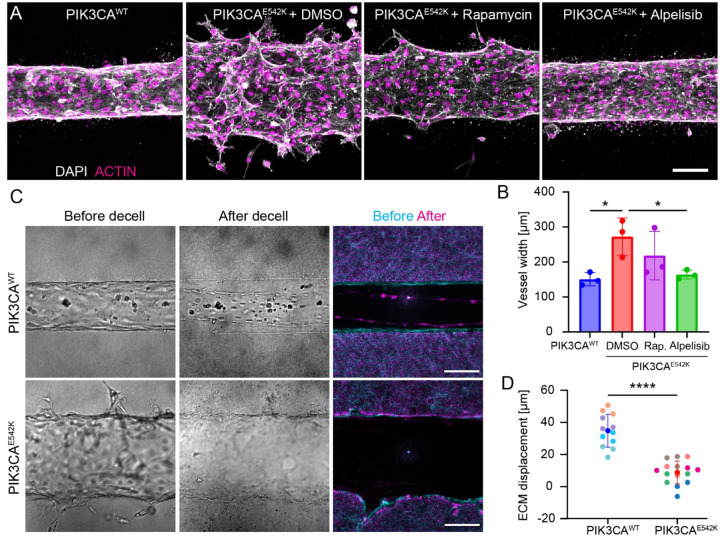
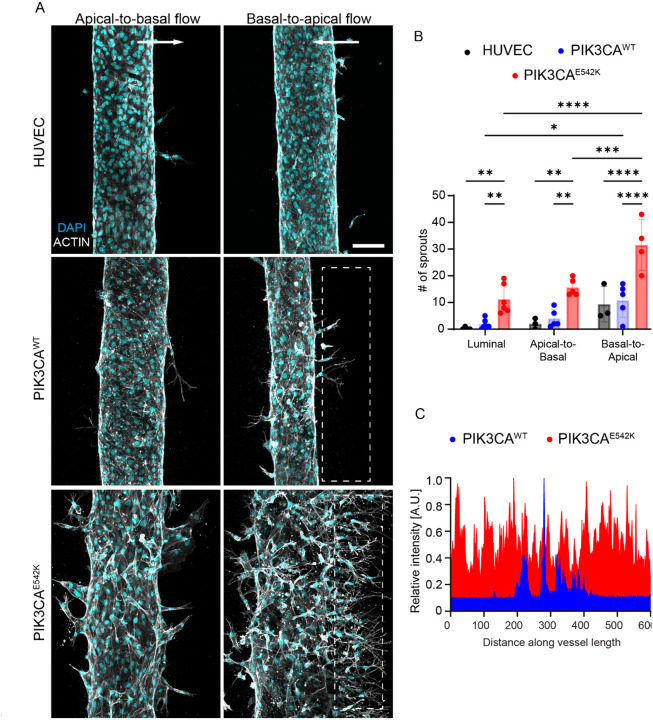
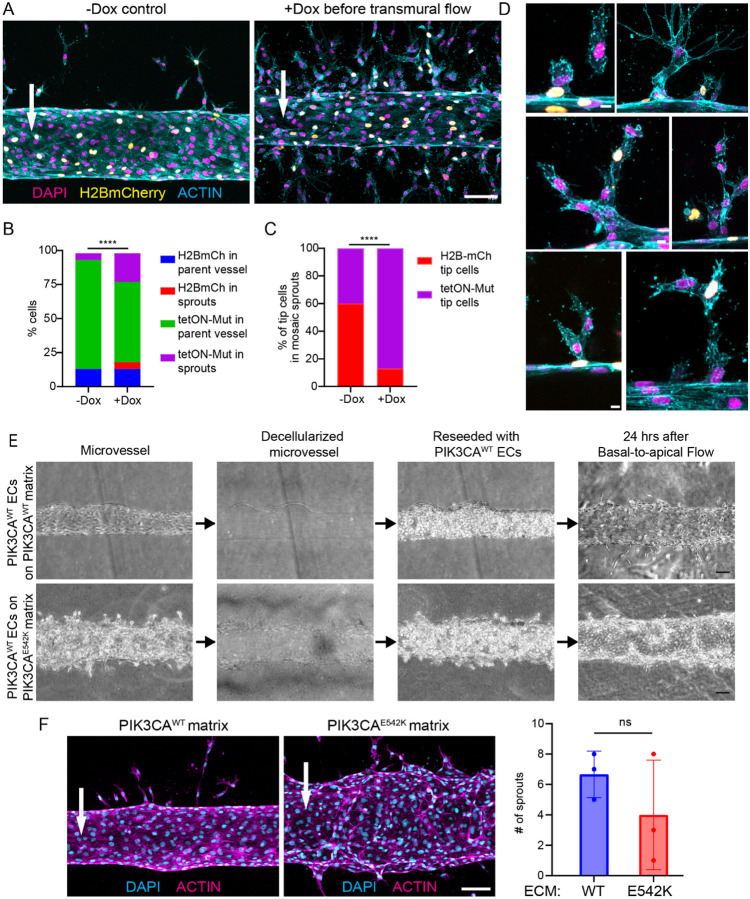
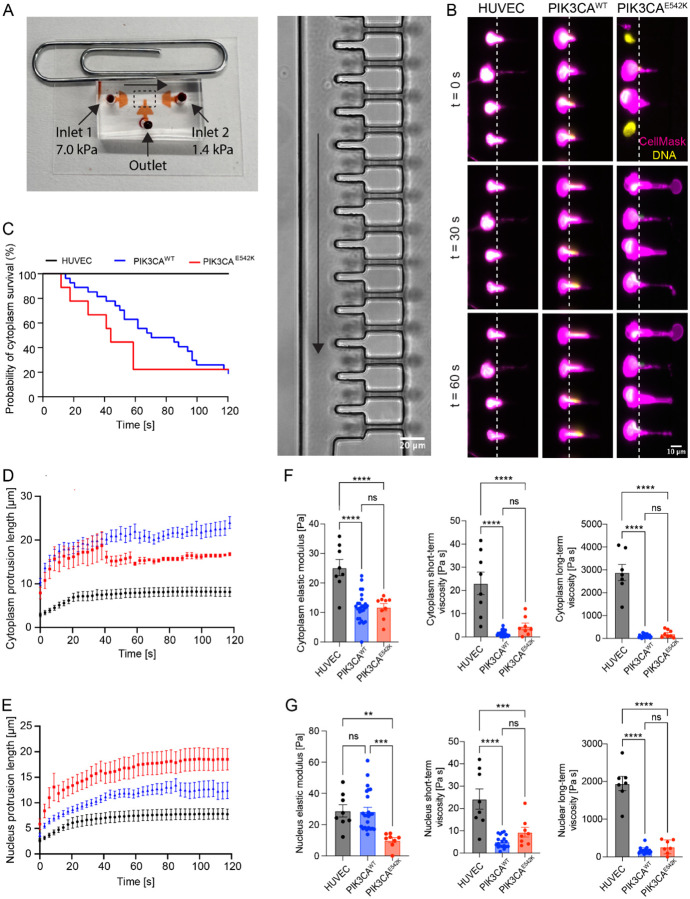
Somatic activating mutations in PIK3CA are common drivers of vascular and lymphatic malformations. Despite common biophysical signatures of tissues susceptible to lesion formation, including compliant extracellular matrix and low rates of perfusion, lesions vary in clinical presentation from localized cystic dilatation to diffuse and infiltrative vascular dysplasia. The mechanisms driving the differences in disease severity and variability in clinical presentation and the role of the biophysical microenvironment in potentiating progression are poorly understood. Here, we investigate the role of hemodynamic forces and the biophysical microenvironment in the pathophysiology of vascular malformations, and we identify hemodynamic shear stress and defective endothelial cell mechanotransduction as key regulators of lesion progression. We found that constitutive PI3K activation impaired flow-mediated endothelial cell alignment and barrier function. We show that defective shear stress sensing in PIK3CA E542K endothelial cells is associated with reduced myosin light chain phosphorylation, junctional instability, and defective recruitment of vinculin to cell-cell junctions. Using 3D microfluidic models of the vasculature, we demonstrate that PIK3CA E542K microvessels apply reduced traction forces and are unaffected by flow interruption. We further found that draining transmural flow resulted in increased sprouting and invasion responses in PIK3CA E542K microvessels. Mechanistically, constitutive PI3K activation decreased cellular and nuclear elasticity resulting in defective cellular tensional homeostasis in endothelial cells which may underlie vascular dilation, tissue hyperplasia, and hypersprouting in PIK3CA-driven venous and lymphatic malformations. Together, these results suggest that defective nuclear mechanics, impaired cellular mechanotransduction, and maladaptive hemodynamic responses contribute to the development and progression of PIK3CA-driven vascular malformations.
Keywords: PIK3CA; Vascular malformations; hemodynamics; mechanotransduction; organs-on-chip; transmural flow; vascular tissue engineering.
Conflict of interest statement
Conflict of Interest The authors have no conflicts to disclose.
Figures


References
-
- Sadick M., Muller-Wille R., Wildgruber M., Wohlgemuth W.A., Vascular Anomalies (Part I): Classification and Diagnostics of Vascular Anomalies, Rofo 190(9) (2018) 825–835. - PubMed
-
- Queisser A., Seront E., Boon L.M., Vikkula M., Genetic Basis and Therapies for Vascular Anomalies, Circ Res 129(1) (2021) 155–173. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous