

This is a preprint.
Cell states and neighborhoods in distinct clinical stages of primary and metastatic esophageal adenocarcinoma
- PMID: 39229240
- PMCID: PMC11370330
- DOI: 10.1101/2024.08.17.608386
Cell states and neighborhoods in distinct clinical stages of primary and metastatic esophageal adenocarcinoma
Update in
-
Cell states and neighborhoods in distinct clinical stages of primary and metastatic esophageal adenocarcinoma.Cell Rep Med. 2025 Jun 17;6(6):102188. doi: 10.1016/j.xcrm.2025.102188. Epub 2025 Jun 10. Cell Rep Med. 2025. PMID: 40499545 Free PMC article.
Abstract
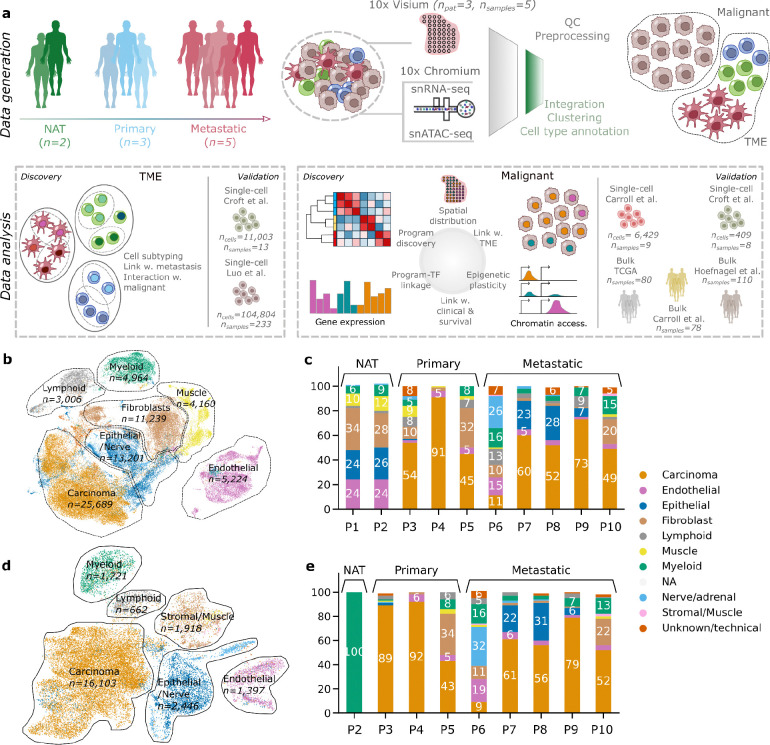
Esophageal adenocarcinoma (EAC) is a highly lethal cancer of the upper gastrointestinal tract with rising incidence in western populations. To decipher EAC disease progression and therapeutic response, we performed multiomic analyses of a cohort of primary and metastatic EAC tumors, incorporating single-nuclei transcriptomic and chromatin accessibility sequencing, along with spatial profiling. We identified tumor microenvironmental features previously described to associate with therapy response. We identified five malignant cell programs, including undifferentiated, intermediate, differentiated, epithelial-to-mesenchymal transition, and cycling programs, which were associated with differential epigenetic plasticity and clinical outcomes, and for which we inferred candidate transcription factor regulons. Furthermore, we revealed diverse spatial localizations of malignant cells expressing their associated transcriptional programs and predicted their significant interactions with microenvironmental cell types. We validated our findings in three external single-cell RNA-seq and three bulk RNA-seq studies. Altogether, our findings advance the understanding of EAC heterogeneity, disease progression, and therapeutic response.
Conflict of interest statement
Declarations of interest E.M.V.: Advisory/Consulting: Enara Bio, Manifold Bio, Monte Rosa, Novartis Institute for Biomedical Research, Serinus Bio, TracerDx Research support: Novartis, BMS, Sanofi, NextPoint Equity: Tango Therapeutics, Genome Medical, Genomic Life, Enara Bio, Manifold Bio, Microsoft, Monte Rosa, Riva Therapeutics, Serinus Bio, Syapse, TracerDx Travel reimbursement: None Patents: Institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation; intermittent legal consulting on patents for Foaley & Hoag Editorial Boards: Science Advances A.J.A. has consulted for Anji Pharmaceuticals, Affini-T Therapeutics, Arrakis Therapeutics, AstraZeneca, Boehringer Ingelheim, Kestrel Therapeutics, Merck & Co., Inc., Mirati Therapeutics, Nimbus Therapeutics, Oncorus, Inc., Plexium, Quanta Therapeutics, Revolution Medicines, Reactive Biosciences, Riva Therapeutics, Servier Pharmaceuticals, Syros Pharmaceuticals, T-knife Therapeutics, Third Rock Ventures, and Ventus Therapeutics. A.J.A. holds equity in Riva Therapeutics and Kestrel Therapeutics. A.J.A. has research funding from Amgen, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Deerfield, Inc., Eli Lilly, Mirati Therapeutics, Nimbus Therapeutics, Novartis, Novo Ventures, Revolution Medicines, and Syros Pharmaceuticals.
Figures







References
-
- Nowicki-Osuch K., Zhuang L., Jammula S., Bleaney C.W., Mahbubani K.T., Devonshire G., Katz-Summercorn A., Eling N., Wilbrey-Clark A., Madissoon E., et al. (2021). Molecular phenotyping reveals the identity of Barrett’s esophagus and its malignant transition. Science 373, 760–767. - PubMed
-
- Pennathur A., Gibson M.K., Jobe B.A., and Luketich J.D. (2013). Oesophageal carcinoma. Lancet 381, 400–412. - PubMed
-
- Derakhshan M.H., Arnold M., Brewster D.H., Going J.J., Mitchell D.R., Forman D., and McColl K.E.L. (2016). Worldwide Inverse Association between Gastric Cancer and Esophageal Adenocarcinoma Suggesting a Common Environmental Factor Exerting Opposing Effects. Am. J. Gastroenterol. 111, 228–239. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources