

C9orf72 dipeptides activate the NLRP3 inflammasome
- PMID: 39229486
- PMCID: PMC11369816
- DOI: 10.1093/braincomms/fcae282
C9orf72 dipeptides activate the NLRP3 inflammasome
Abstract
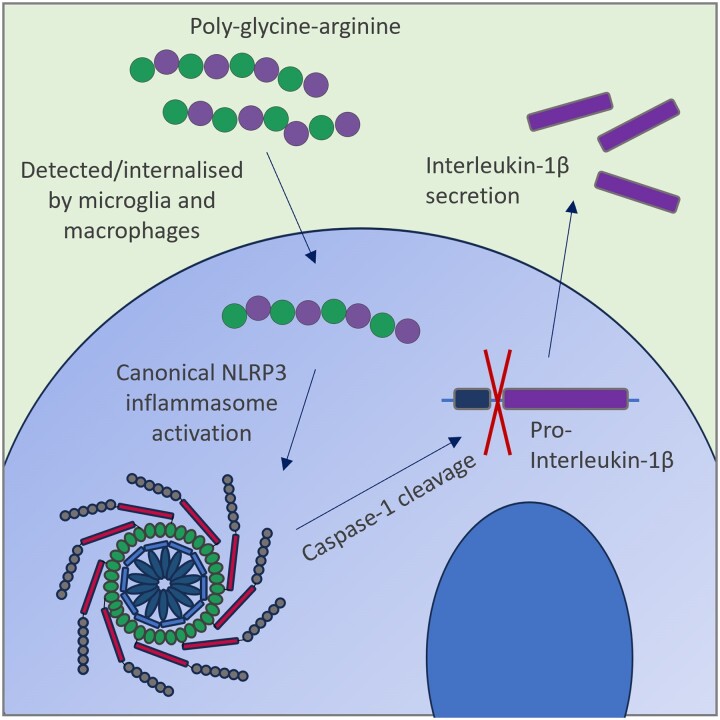
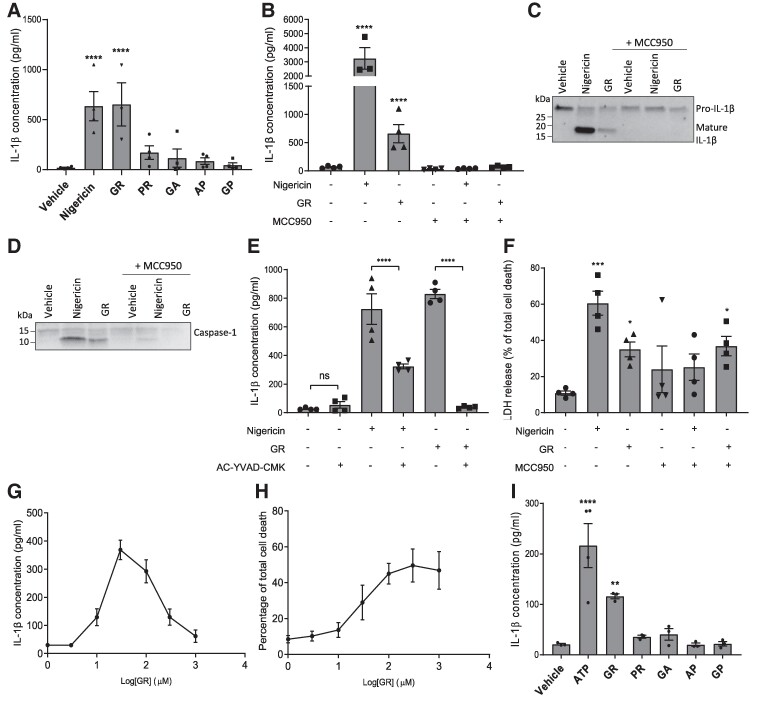
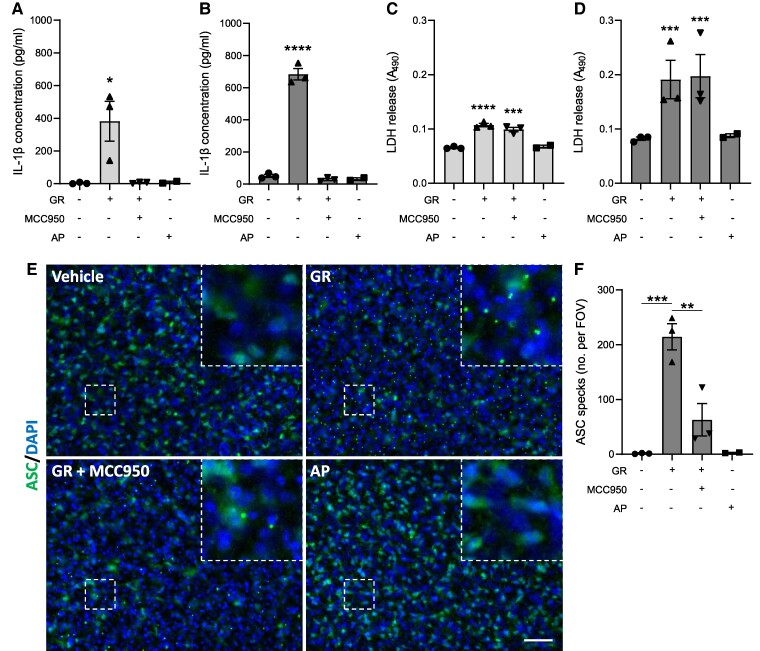
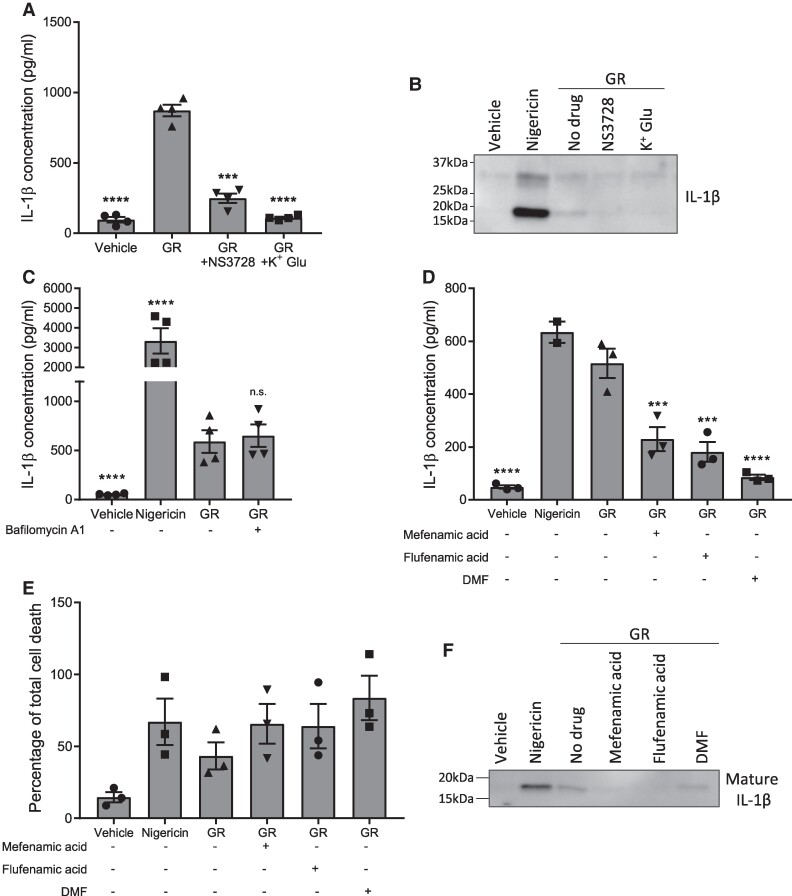
Frontotemporal dementia and amyotrophic lateral sclerosis are neurodegenerative diseases with considerable clinical, genetic and pathological overlap. The most common cause of both diseases is a hexanucleotide repeat expansion in C9orf72. The expansion is translated to produce five toxic dipeptides, which aggregate in patient brain. Neuroinflammation is a feature of frontotemporal dementia and amyotrophic lateral sclerosis; however, its causes are unknown. The nod-like receptor family, pyrin domain-containing 3 inflammasome is implicated in several other neurodegenerative diseases as a driver of damaging inflammation. The inflammasome is a multi-protein complex which forms in immune cells in response to tissue damage, pathogens or aggregating proteins. Inflammasome activation is observed in models of other neurodegenerative diseases such as Alzheimer's disease, and inflammasome inhibition rescues cognitive decline in rodent models of Alzheimer's disease. Here, we show that a dipeptide arising from the C9orf72 expansion, poly-glycine-arginine, activated the inflammasome in microglia and macrophages, leading to secretion of the pro-inflammatory cytokine, interleukin-1β. Poly-glycine-arginine also activated the inflammasome in organotypic hippocampal slice cultures, and immunofluorescence imaging demonstrated formation of inflammasome specks in response to poly-glycine-arginine. Several clinically available anti-inflammatory drugs rescued poly-glycine-arginine-induced inflammasome activation. These data suggest that C9orf72 dipeptides contribute to the neuroinflammation observed in patients, and highlight the inflammasome as a potential therapeutic target for frontotemporal dementia and amyotrophic lateral sclerosis.
Keywords: ALS; NLRP3; c9orf72; frontotemporal dementia; inflammasome.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors have no competing interests to declare.
Figures
References
-
- Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous