

Single-molecule digital sizing of proteins in solution
- PMID: 39231922
- PMCID: PMC11375031
- DOI: 10.1038/s41467-024-50825-9
Single-molecule digital sizing of proteins in solution
Abstract
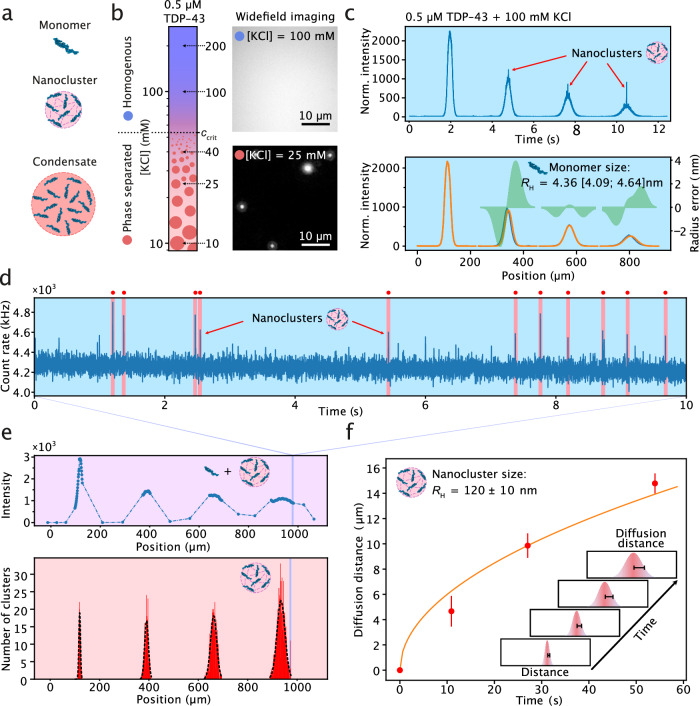
The physical characterization of proteins in terms of their sizes, interactions, and assembly states is key to understanding their biological function and dysfunction. However, this has remained a difficult task because proteins are often highly polydisperse and present as multicomponent mixtures. Here, we address this challenge by introducing single-molecule microfluidic diffusional sizing (smMDS). This approach measures the hydrodynamic radius of single proteins and protein assemblies in microchannels using single-molecule fluorescence detection. smMDS allows for ultrasensitive sizing of proteins down to femtomolar concentrations and enables affinity profiling of protein interactions at the single-molecule level. We show that smMDS is effective in resolving the assembly states of protein oligomers and in characterizing the size of protein species within complex mixtures, including fibrillar protein aggregates and nanoscale condensate clusters. Overall, smMDS is a highly sensitive method for the analysis of proteins in solution, with wide-ranging applications in drug discovery, diagnostics, and nanobiotechnology.
© 2024. The Author(s).
Conflict of interest statement
G.K., K.L.S., W.E.A., and T.P.J.K. declare the following competing interests. Parts of this work have been the subject of a patent application filed by Cambridge Enterprise Limited, a fully-owned subsidiary of the University of Cambridge. Inventors: Krainer, G.; Saar, K.L.; Arter, W.E., Knowles, T.P.J.; Applicant: Cambridge Enterprise Ltd.; Title: Highly sensitive biomolecule detection and quantification. Publication Number: WO/2021/176065; Publication Date: 10.09.2021; International Application No.: PCT/EP2021/055614; International Filing Date: 05.03.2021. The remaining authors declare no competing interests.
Figures







References
-
- Goodsell, D. S. The Machinery of Life. The Machinery of Life (Springer, New York, NY, 2009). 10.1007/978-0-387-84925-6.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
