

DNA methylation controls stemness of astrocytes in health and ischaemia
- PMID: 39232166
- PMCID: PMC11464379
- DOI: 10.1038/s41586-024-07898-9
DNA methylation controls stemness of astrocytes in health and ischaemia
Abstract
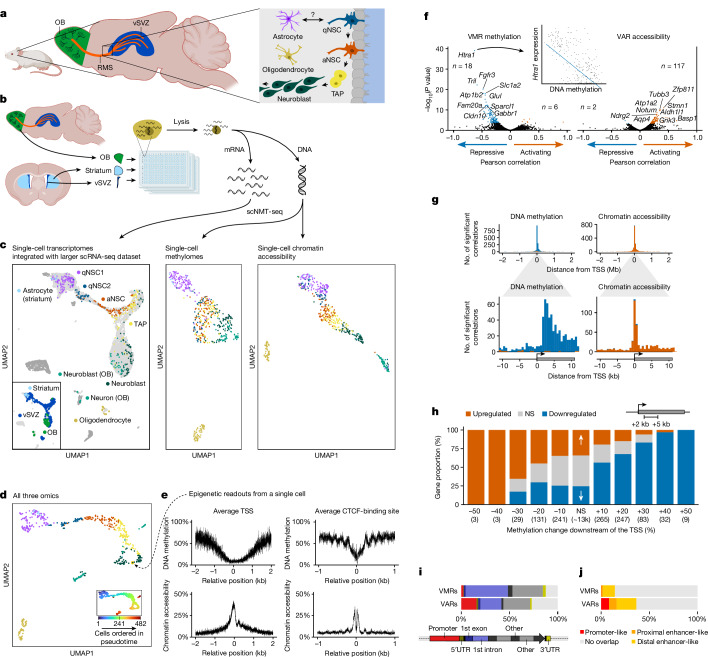
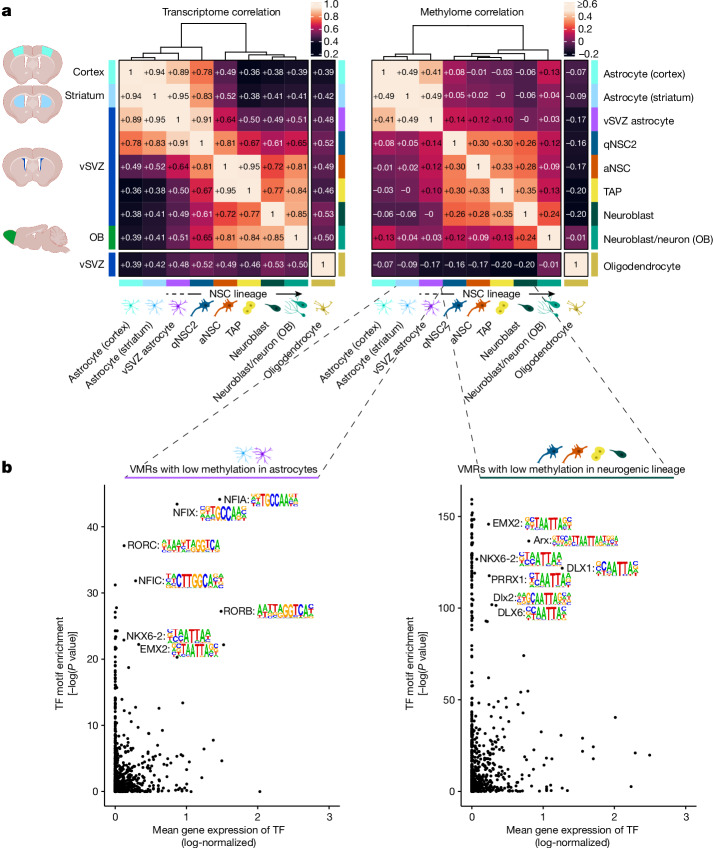
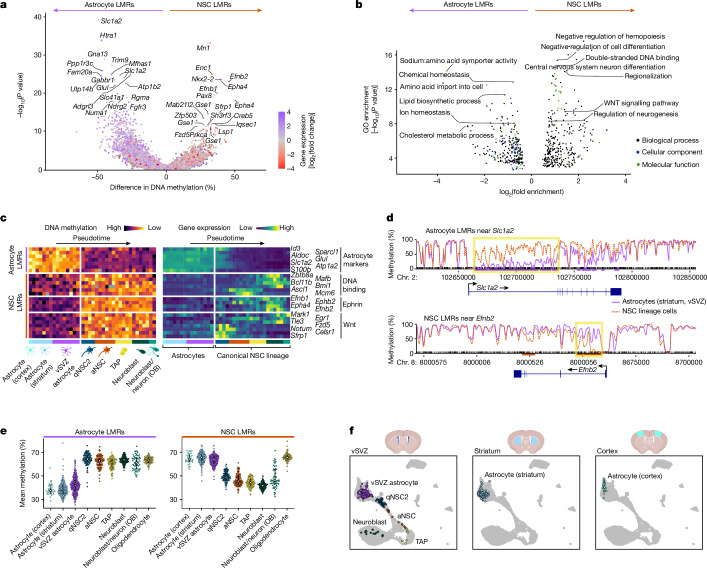
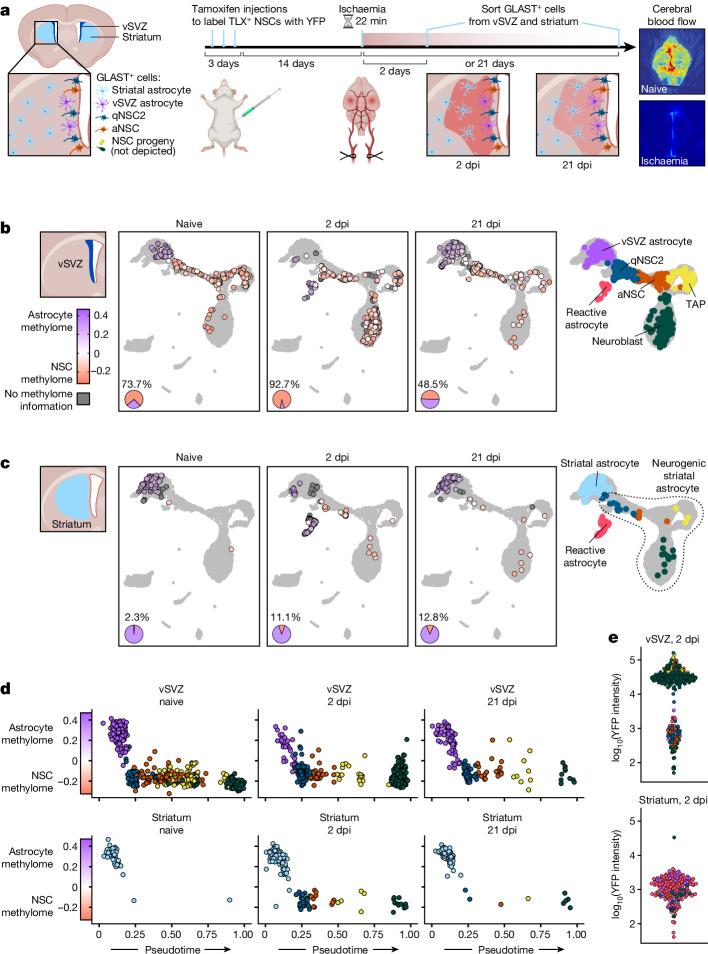
Astrocytes are the most abundant cell type in the mammalian brain and provide structural and metabolic support to neurons, regulate synapses and become reactive after injury and disease. However, a small subset of astrocytes settles in specialized areas of the adult brain where these astrocytes instead actively generate differentiated neuronal and glial progeny and are therefore referred to as neural stem cells1-3. Common parenchymal astrocytes and quiescent neural stem cells share similar transcriptomes despite their very distinct functions4-6. Thus, how stem cell activity is molecularly encoded remains unknown. Here we examine the transcriptome, chromatin accessibility and methylome of neural stem cells and their progeny, and of astrocytes from the striatum and cortex in the healthy and ischaemic adult mouse brain. We identify distinct methylation profiles associated with either astrocyte or stem cell function. Stem cell function is mediated by methylation of astrocyte genes and demethylation of stem cell genes that are expressed later. Ischaemic injury to the brain induces gain of stemness in striatal astrocytes7. We show that this response involves reprogramming the astrocyte methylome to a stem cell methylome and is absent if the de novo methyltransferase DNMT3A is missing. Overall, we unveil DNA methylation as a promising target for regenerative medicine.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures









References
-
- Delgado, A. C. et al. Release of stem cells from quiescence reveals gliogenic domains in the adult mouse brain. Science372, 1205–1209 (2021). - PubMed
-
- Llorens-Bobadilla, E. et al. Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell17, 329–340 (2015). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
