

Mapping glycoprotein structure reveals Flaviviridae evolutionary history
- PMID: 39232167
- PMCID: PMC11410658
- DOI: 10.1038/s41586-024-07899-8
Mapping glycoprotein structure reveals Flaviviridae evolutionary history
Abstract
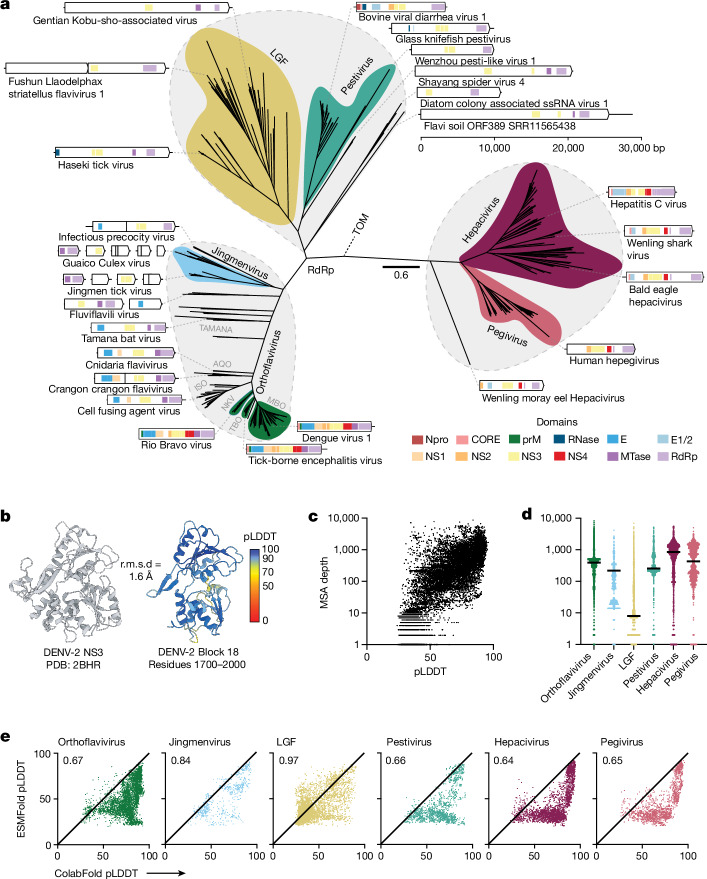
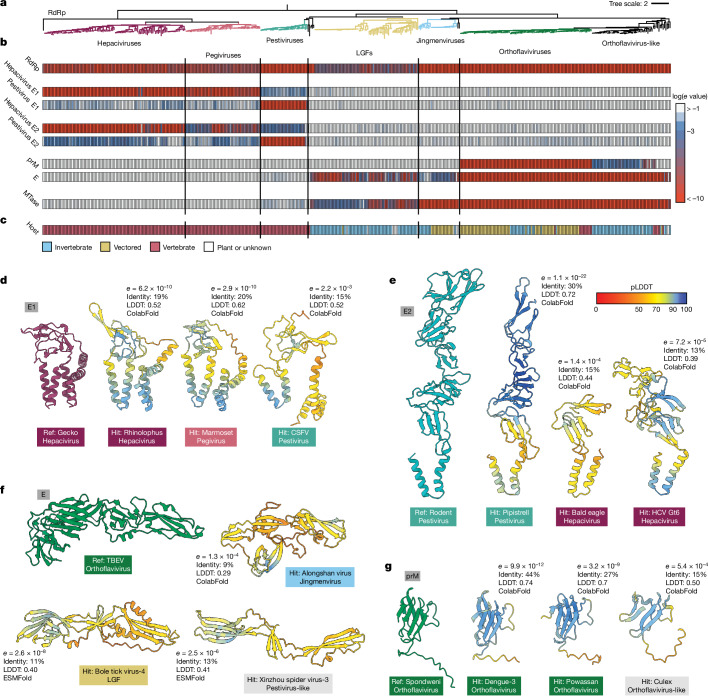
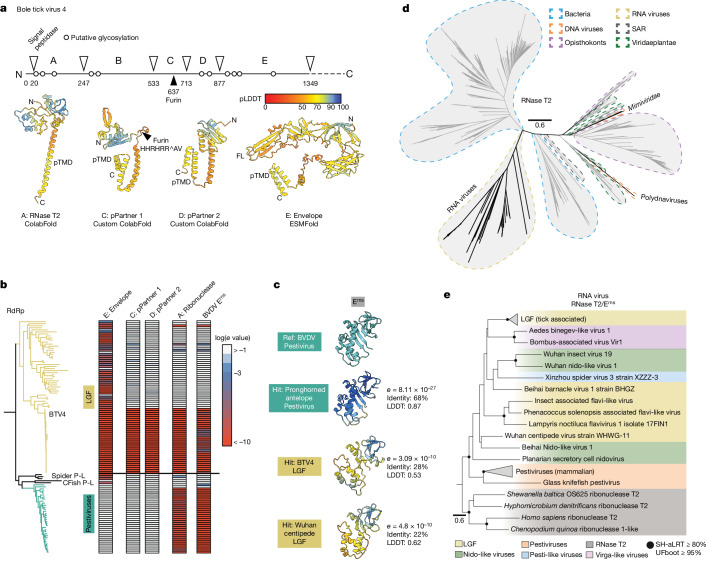
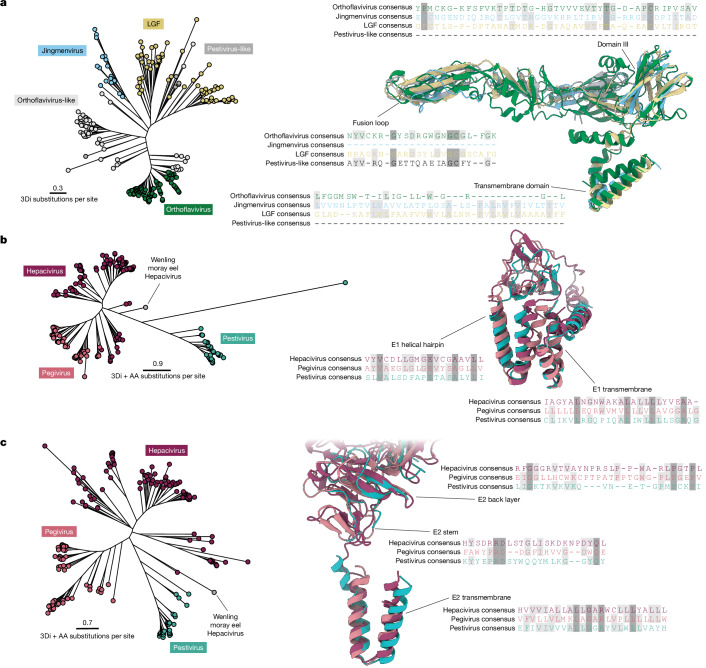
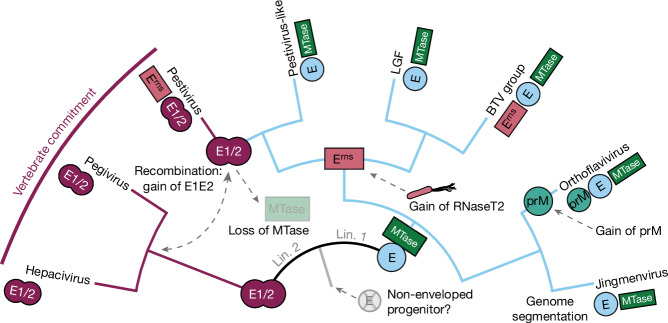
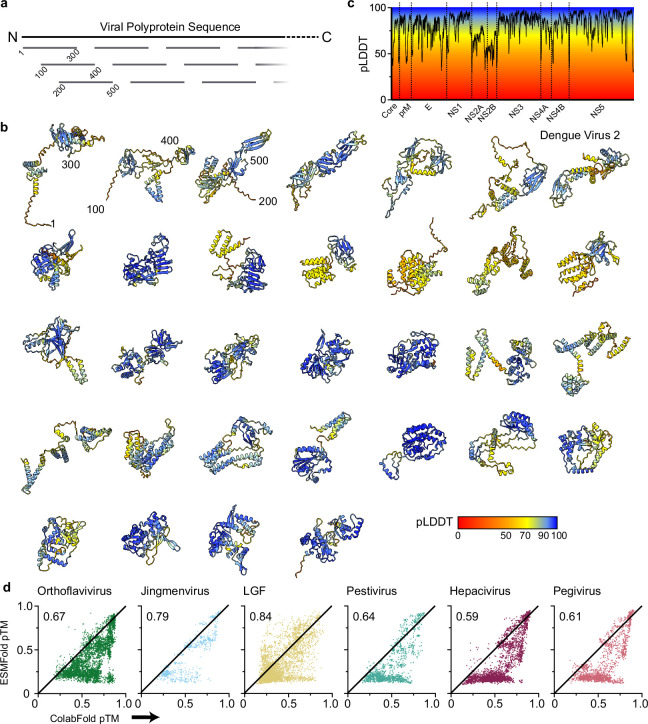
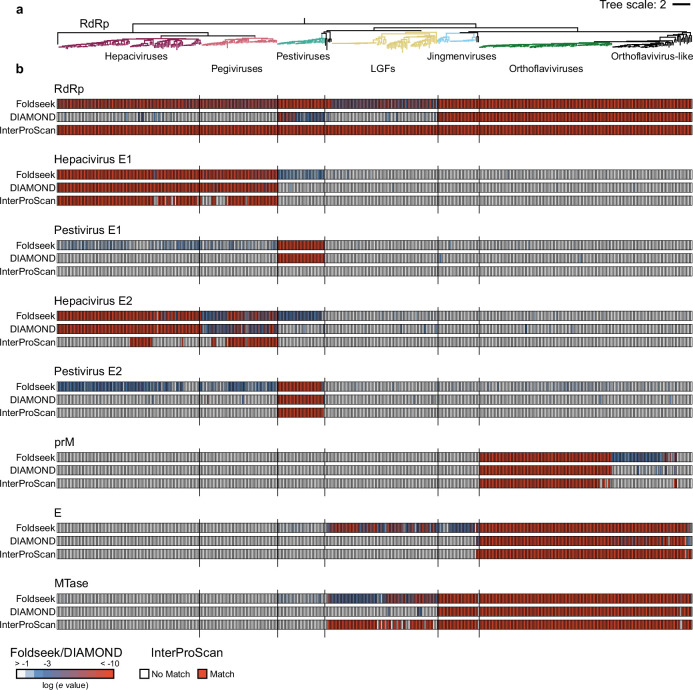
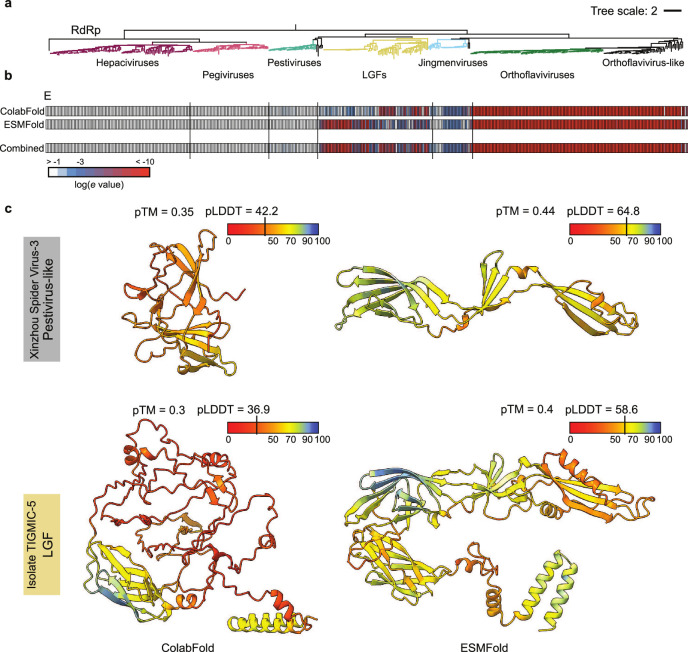
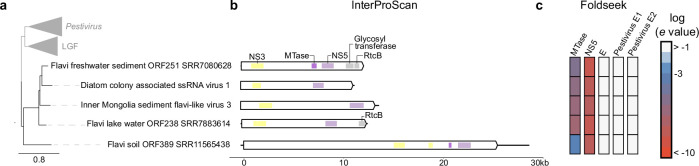
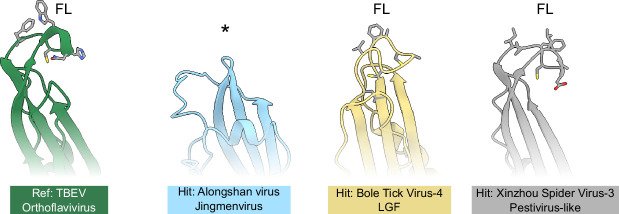
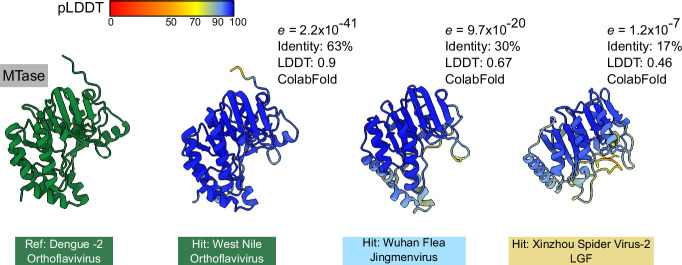
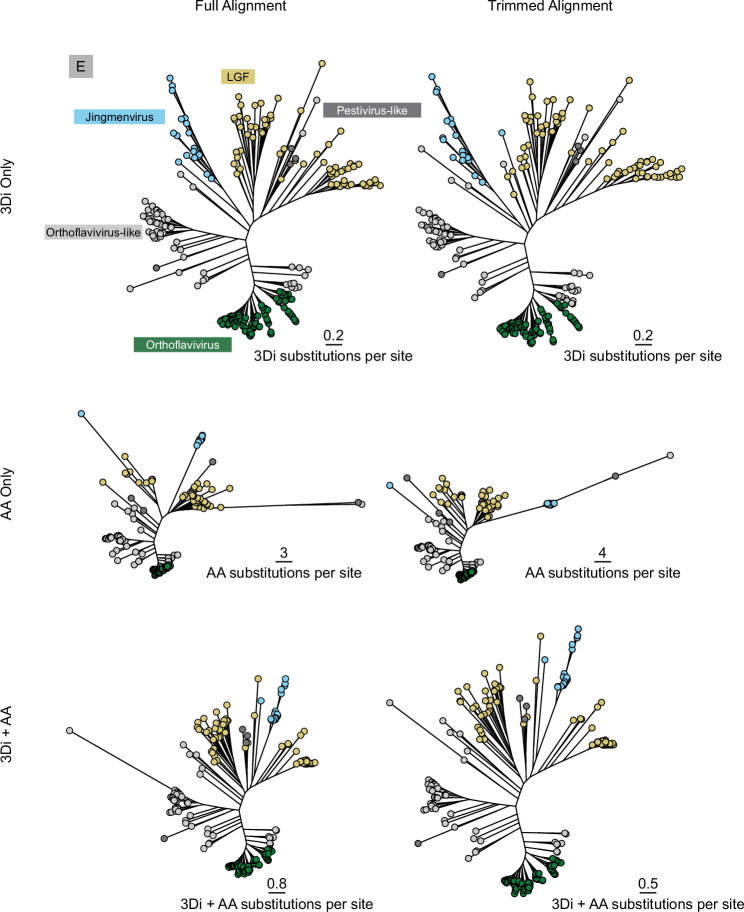
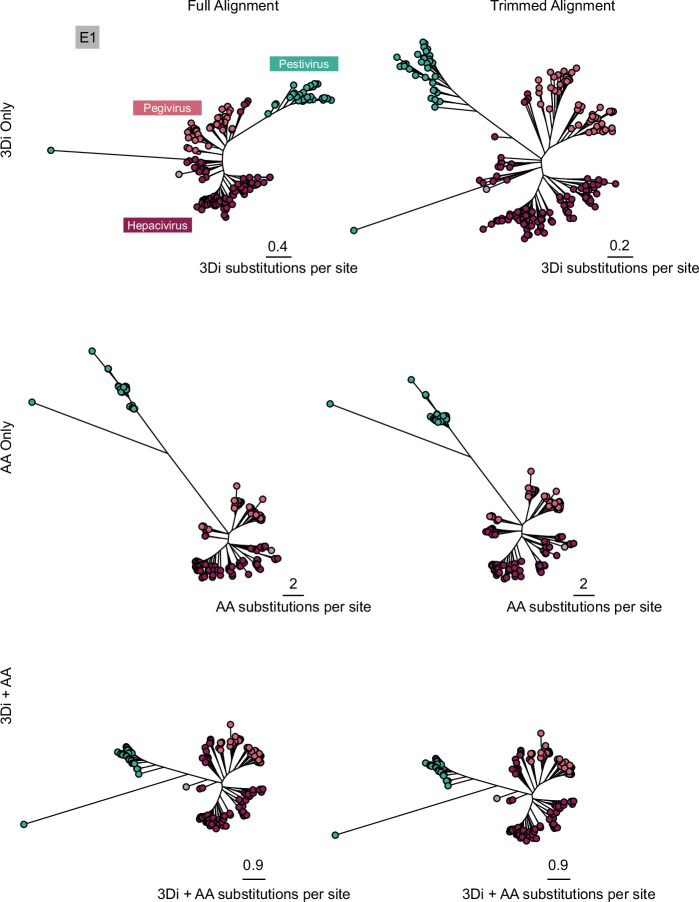
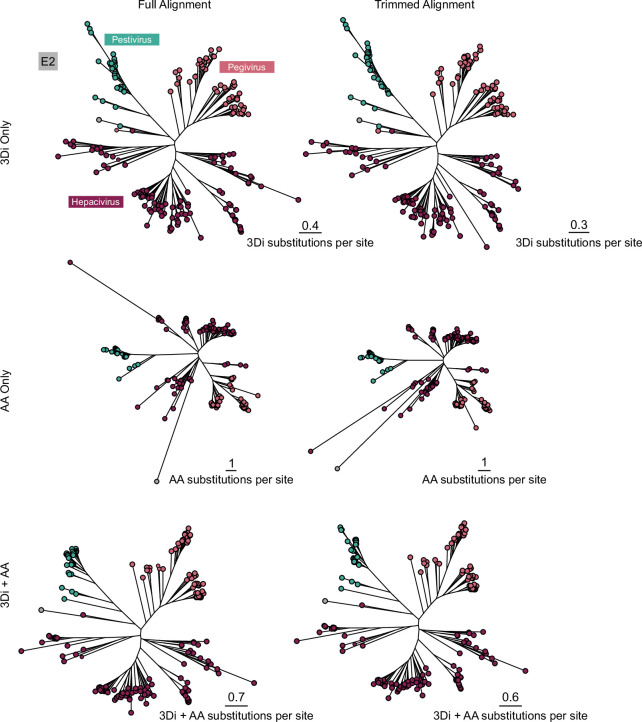
Viral glycoproteins drive membrane fusion in enveloped viruses and determine host range, tissue tropism and pathogenesis1. Despite their importance, there is a fragmentary understanding of glycoproteins within the Flaviviridae2, a large virus family that include pathogens such as hepatitis C, dengue and Zika viruses, and numerous other human, animal and emergent viruses. For many flaviviruses the glycoproteins have not yet been identified, for others, such as the hepaciviruses, the molecular mechanisms of membrane fusion remain uncharacterized3. Here we combine phylogenetic analyses with protein structure prediction to survey glycoproteins across the entire Flaviviridae. We find class II fusion systems, homologous to the Orthoflavivirus E glycoprotein in most species, including highly divergent jingmenviruses and large genome flaviviruses. However, the E1E2 glycoproteins of the hepaciviruses, pegiviruses and pestiviruses are structurally distinct, may represent a novel class of fusion mechanism, and are strictly associated with infection of vertebrate hosts. By mapping glycoprotein distribution onto the underlying phylogeny, we reveal a complex evolutionary history marked by the capture of bacterial genes and potentially inter-genus recombination. These insights, made possible through protein structure prediction, refine our understanding of viral fusion mechanisms and reveal the events that have shaped the diverse virology and ecology of the Flaviviridae.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
