

Farmed fur animals harbour viruses with zoonotic spillover potential
- PMID: 39232170
- PMCID: PMC11741233
- DOI: 10.1038/s41586-024-07901-3
Farmed fur animals harbour viruses with zoonotic spillover potential
Abstract
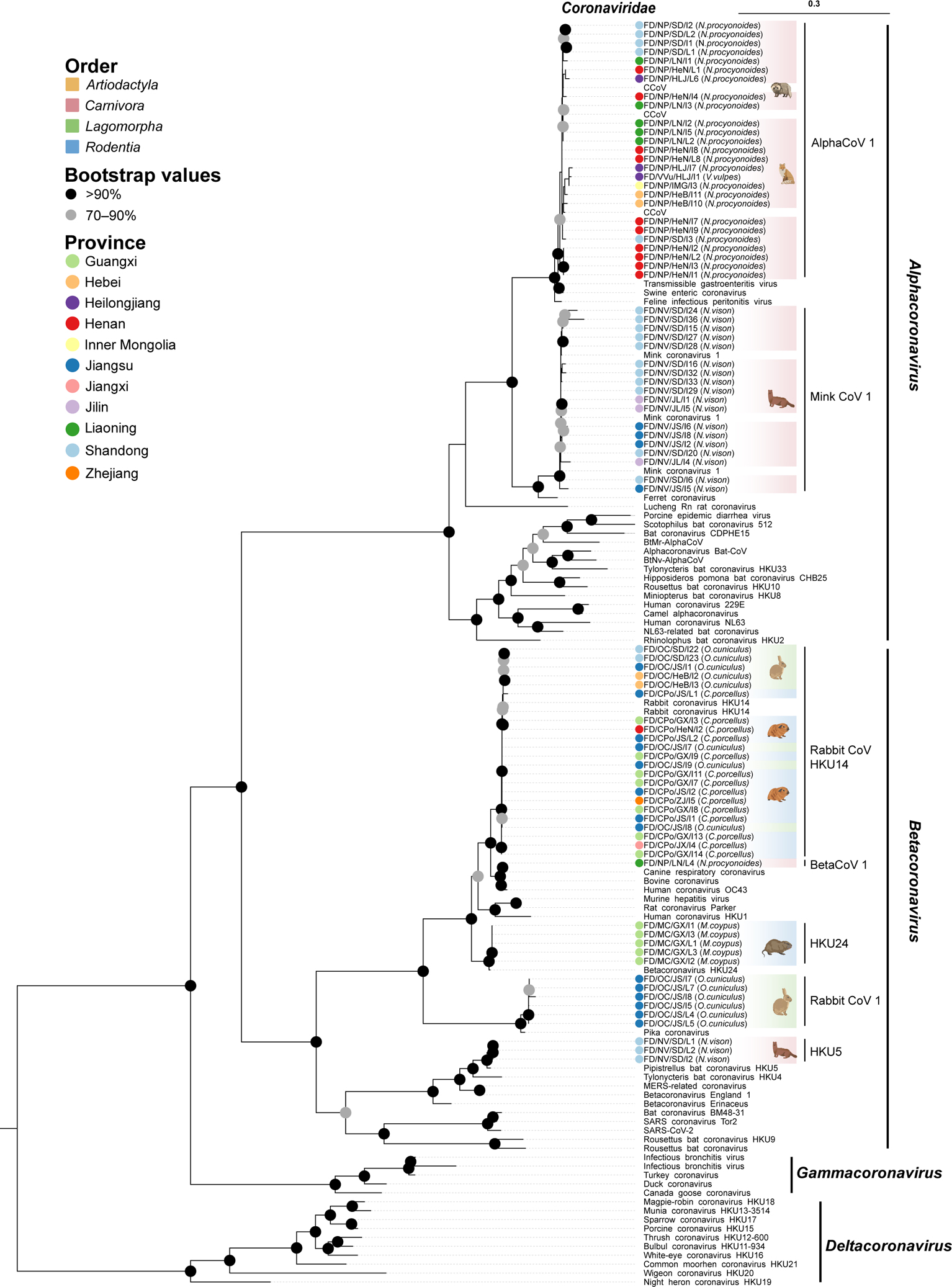
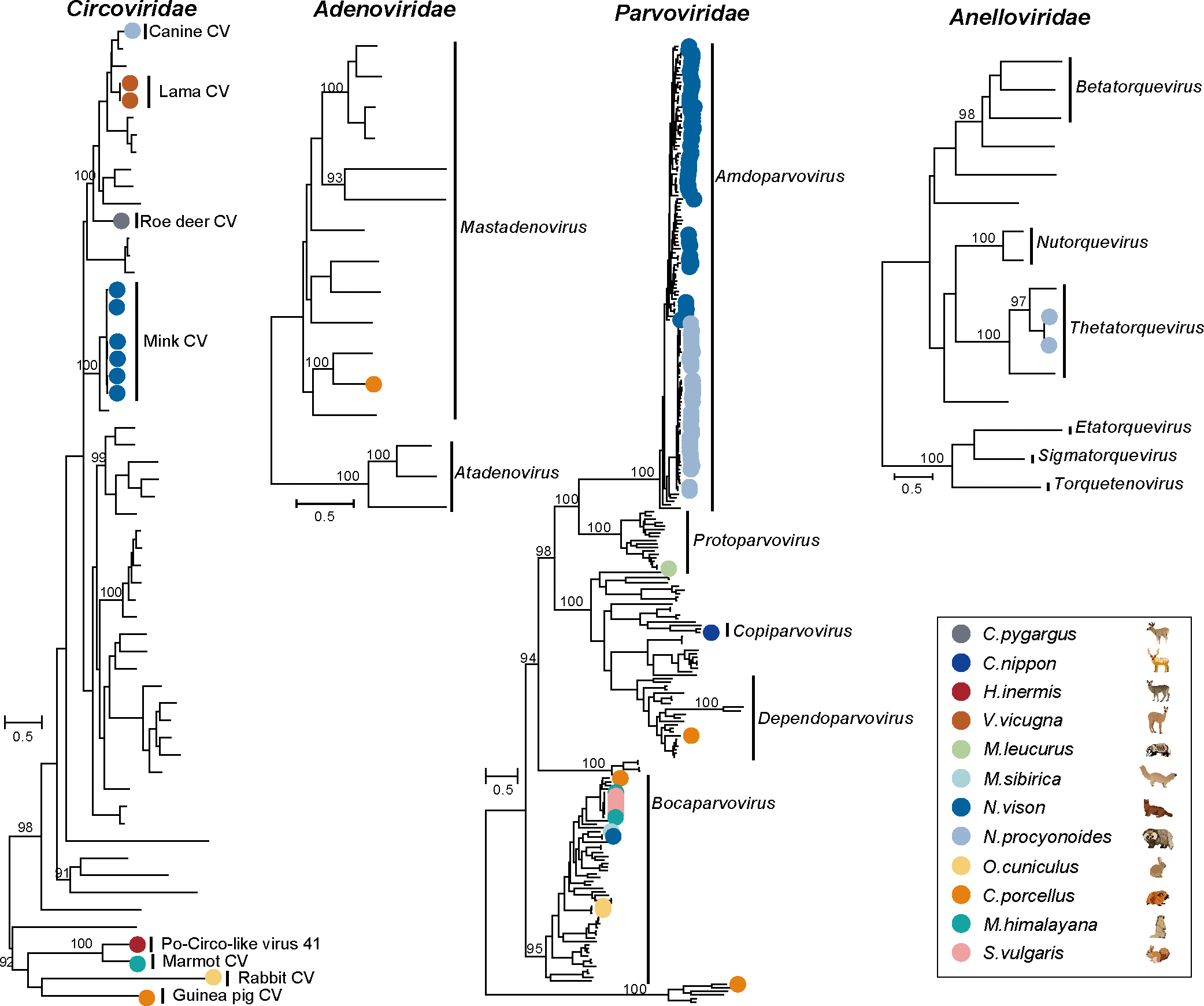
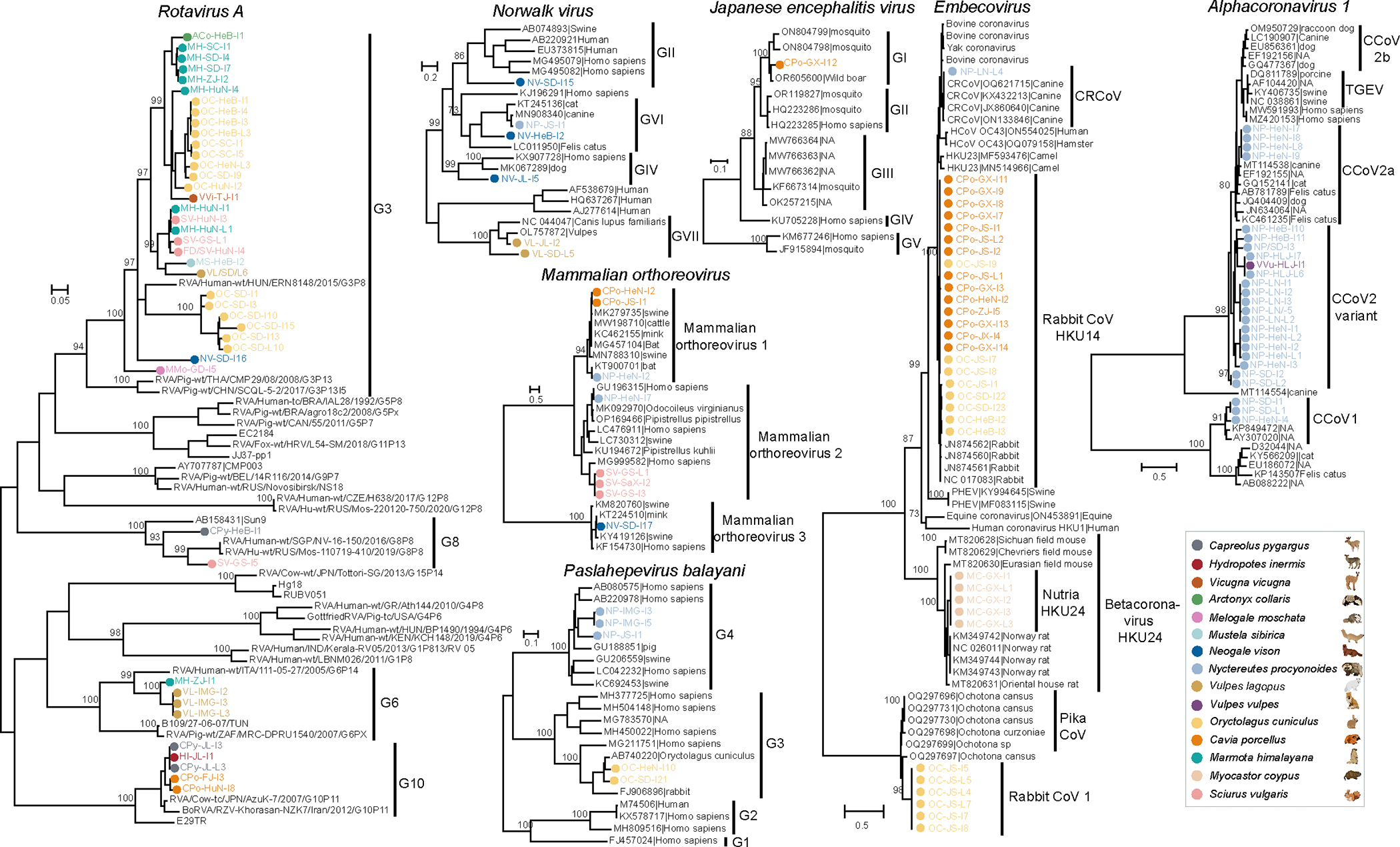
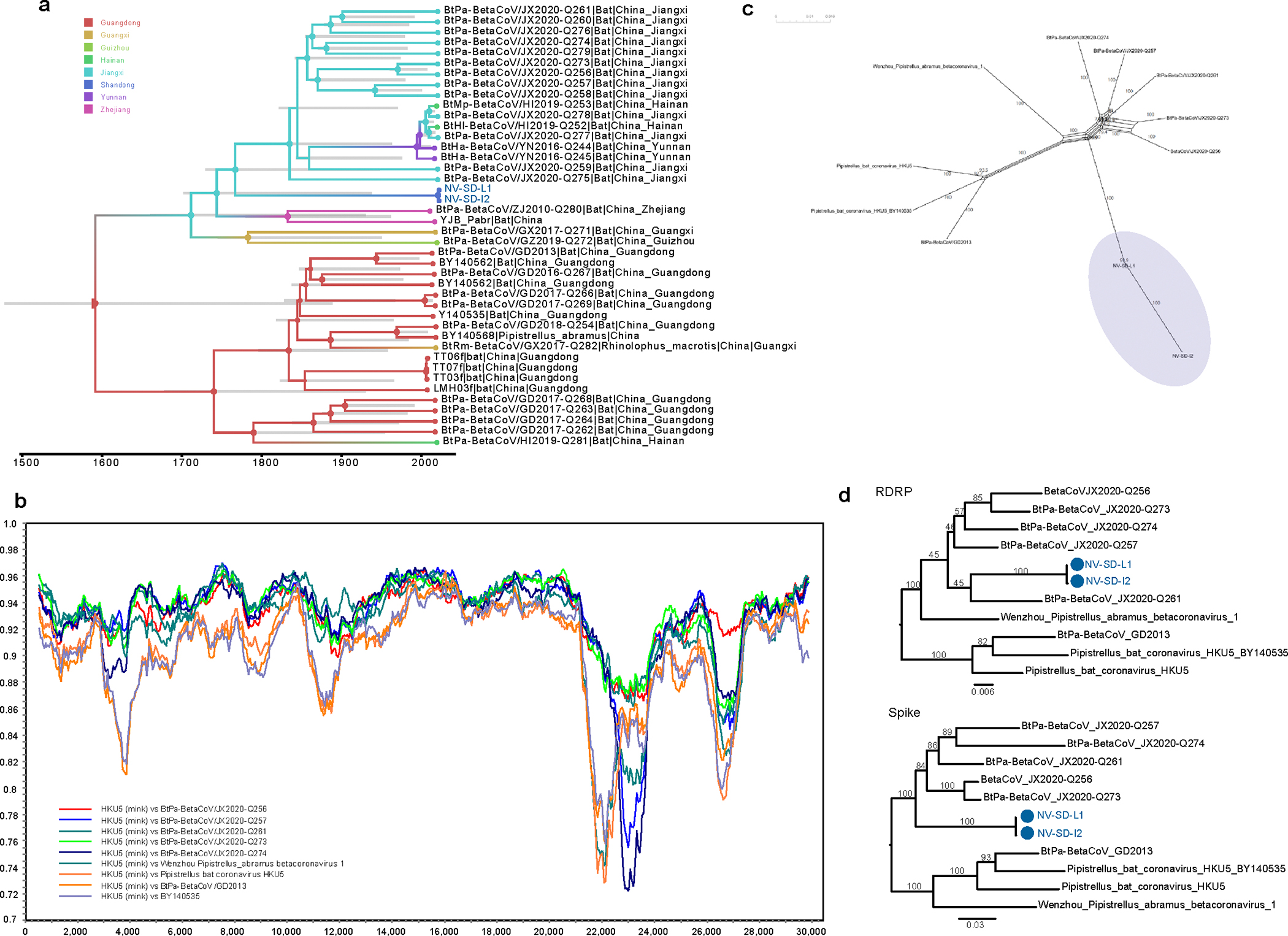
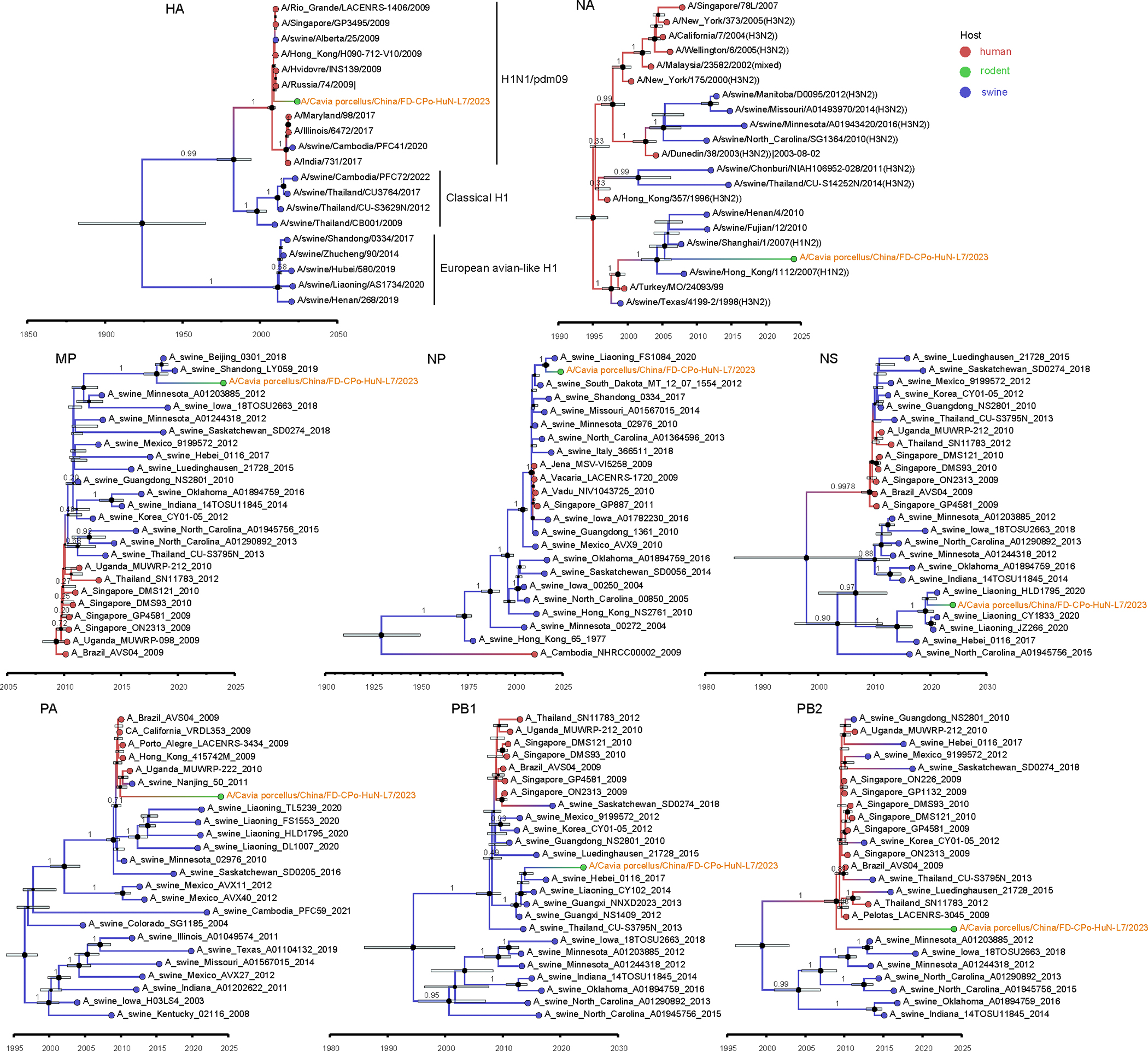
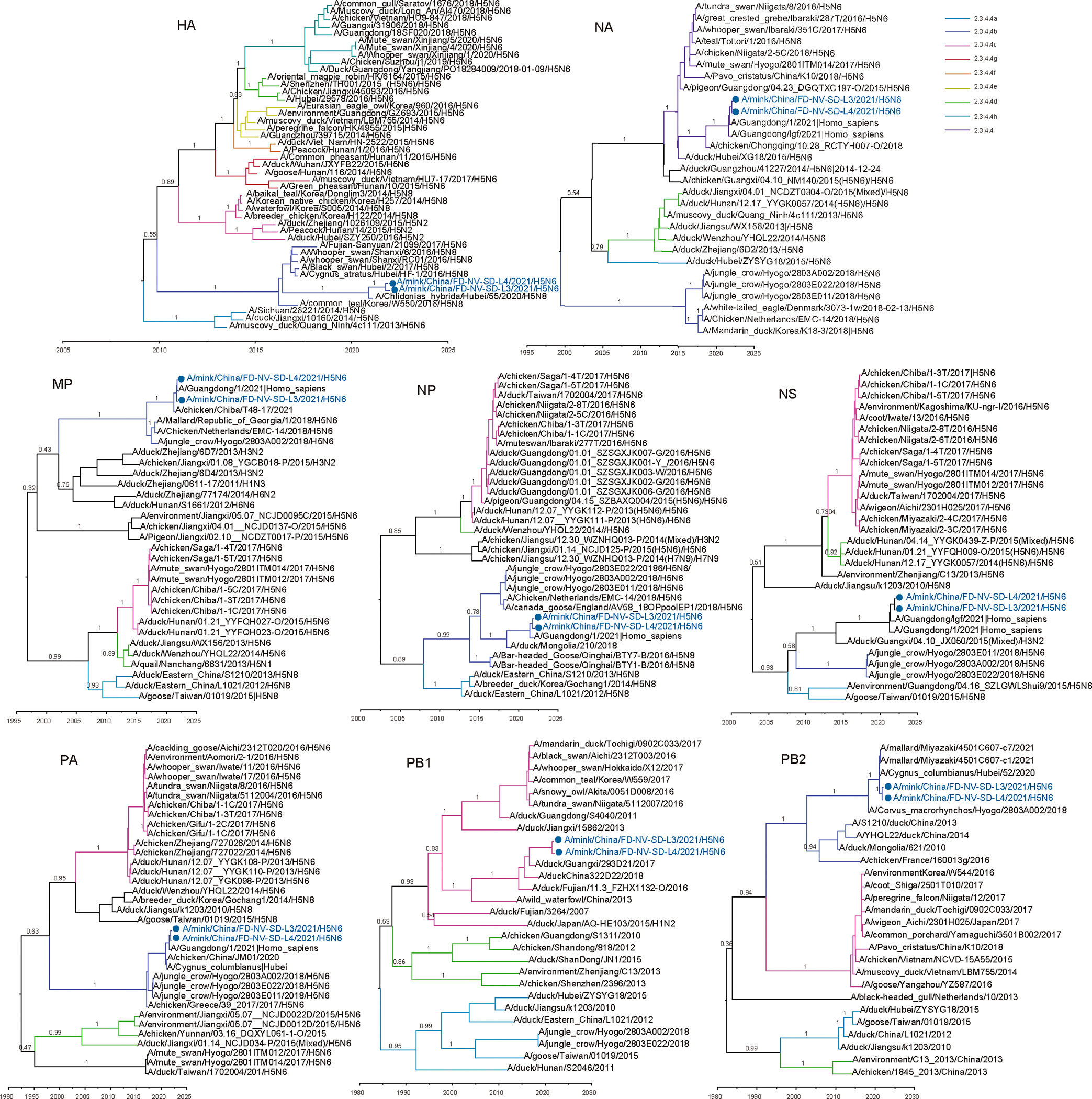
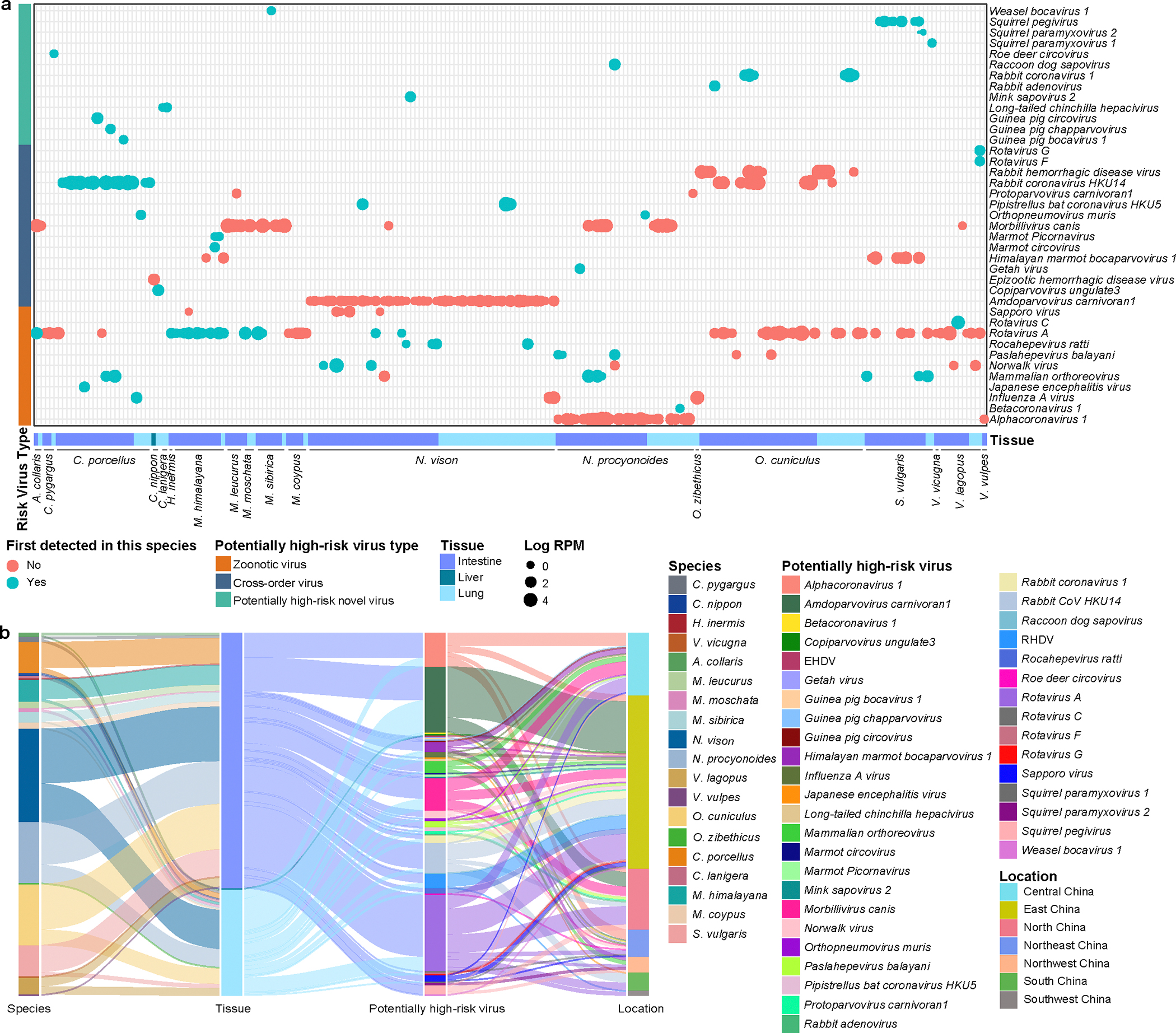
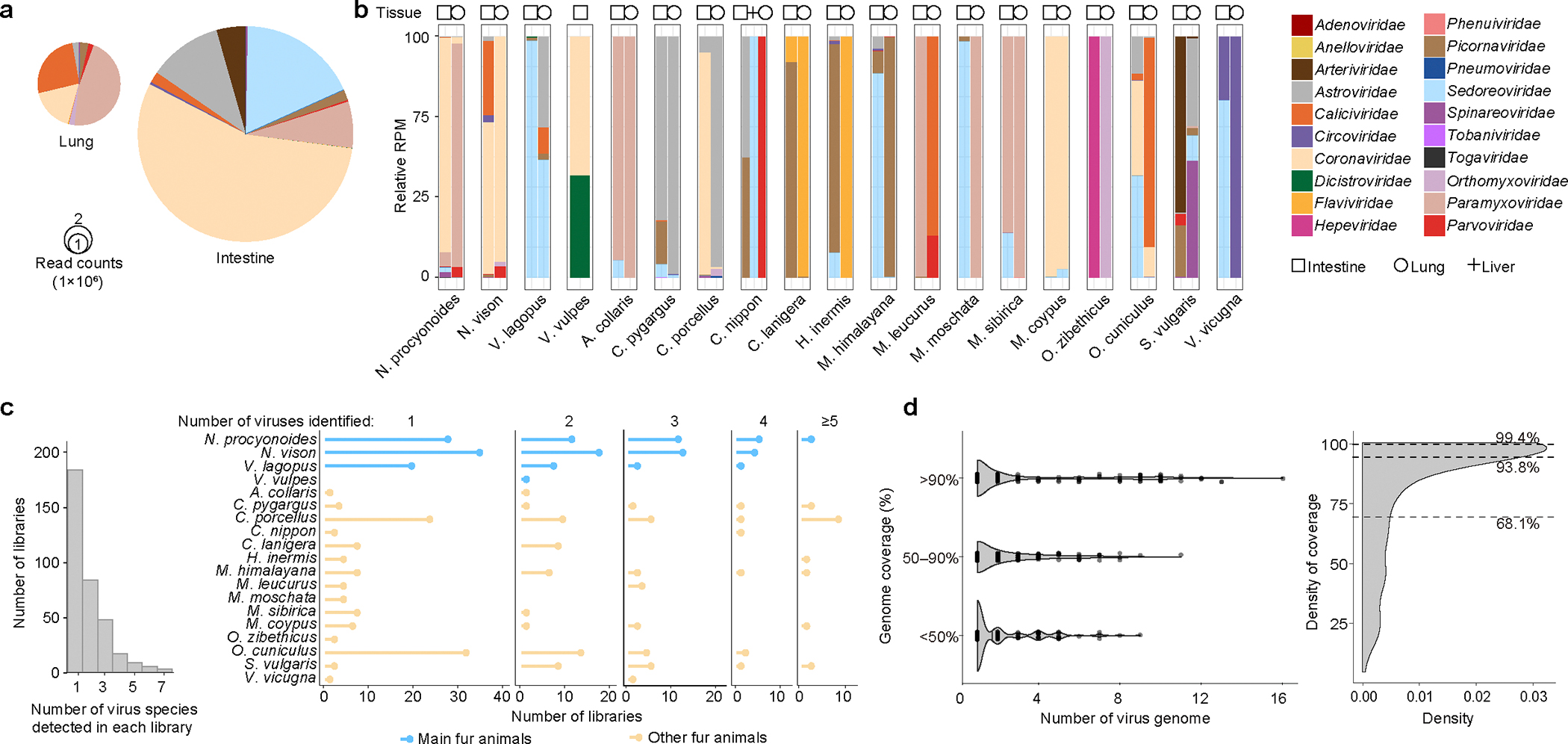
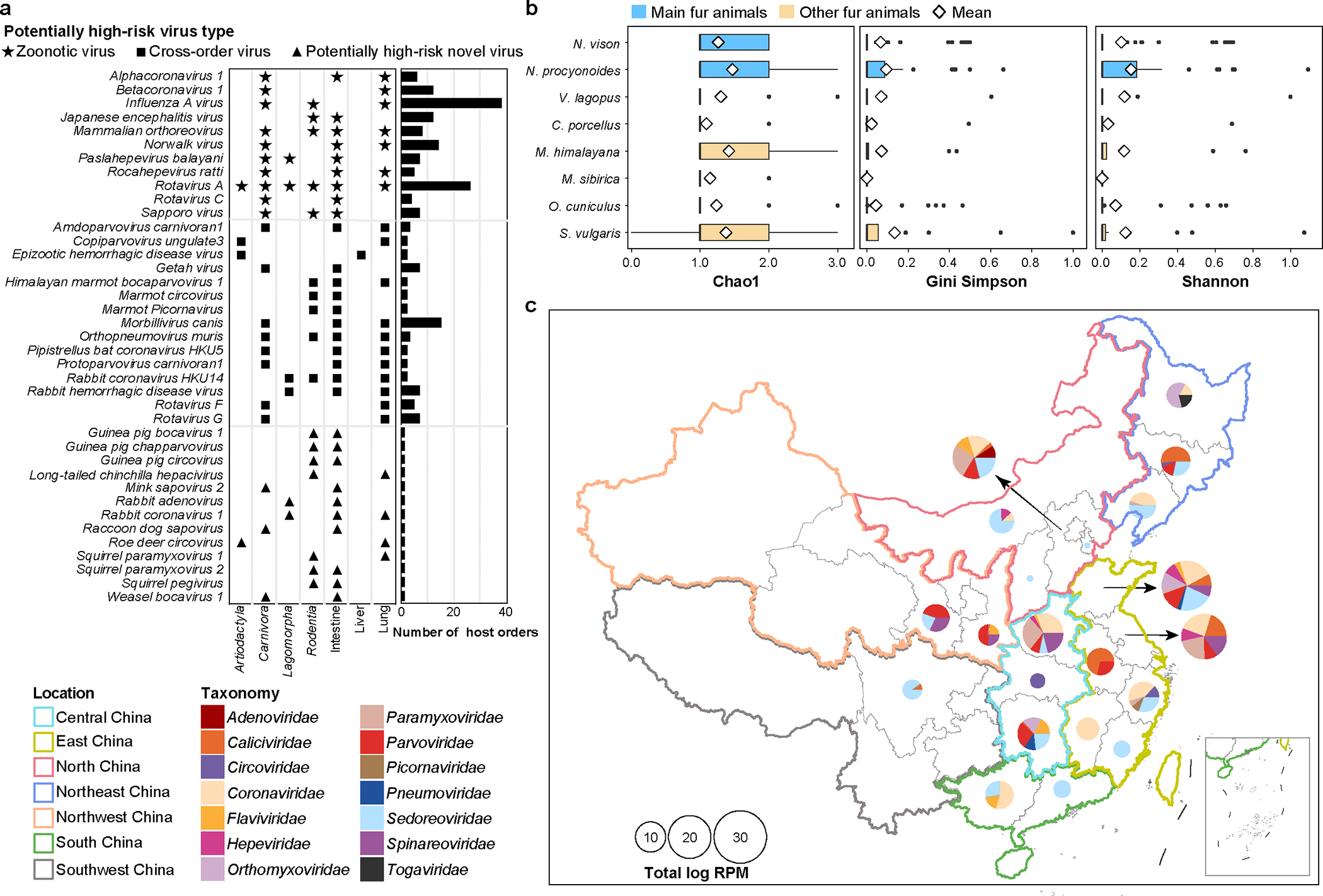
Animals such as raccoon dogs, mink and muskrats are farmed for fur and are sometimes used as food or medicinal products1,2, yet they are also potential reservoirs of emerging pathogens3. Here we performed single-sample metatranscriptomic sequencing of internal tissues from 461 individual fur animals that were found dead due to disease. We characterized 125 virus species, including 36 that were novel and 39 at potentially high risk of cross-species transmission, including zoonotic spillover. Notably, we identified seven species of coronaviruses, expanding their known host range, and documented the cross-species transmission of a novel canine respiratory coronavirus to raccoon dogs and of bat HKU5-like coronaviruses to mink, present at a high abundance in lung tissues. Three subtypes of influenza A virus-H1N2, H5N6 and H6N2-were detected in the lungs of guinea pig, mink and muskrat, respectively. Multiple known zoonotic viruses, such as Japanese encephalitis virus and mammalian orthoreovirus4,5, were detected in guinea pigs. Raccoon dogs and mink carried the highest number of potentially high-risk viruses, while viruses from the Coronaviridae, Paramyxoviridae and Sedoreoviridae families commonly infected multiple hosts. These data also reveal potential virus transmission between farmed animals and wild animals, and from humans to farmed animals, indicating that fur farming represents an important transmission hub for viral zoonoses.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
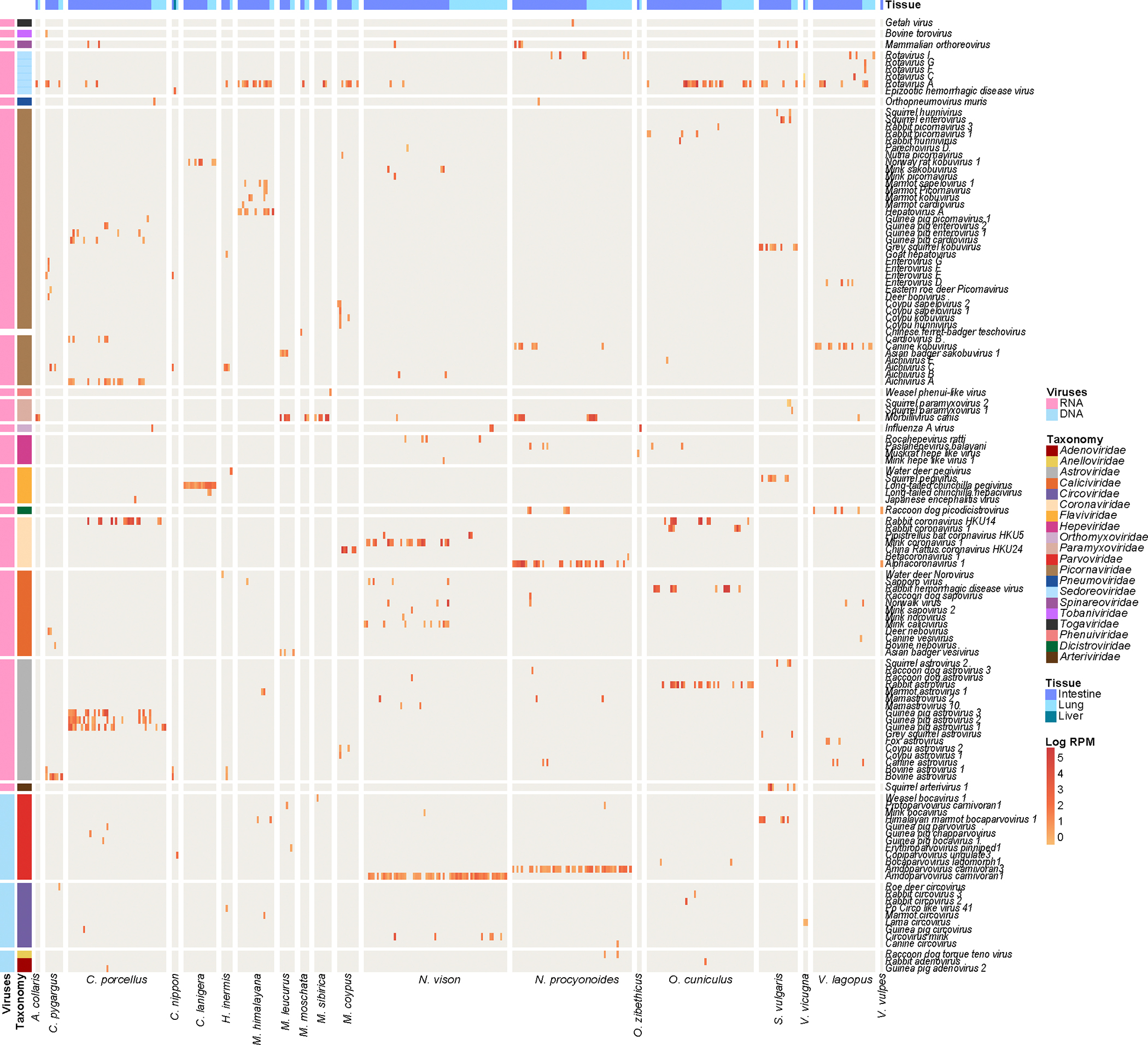
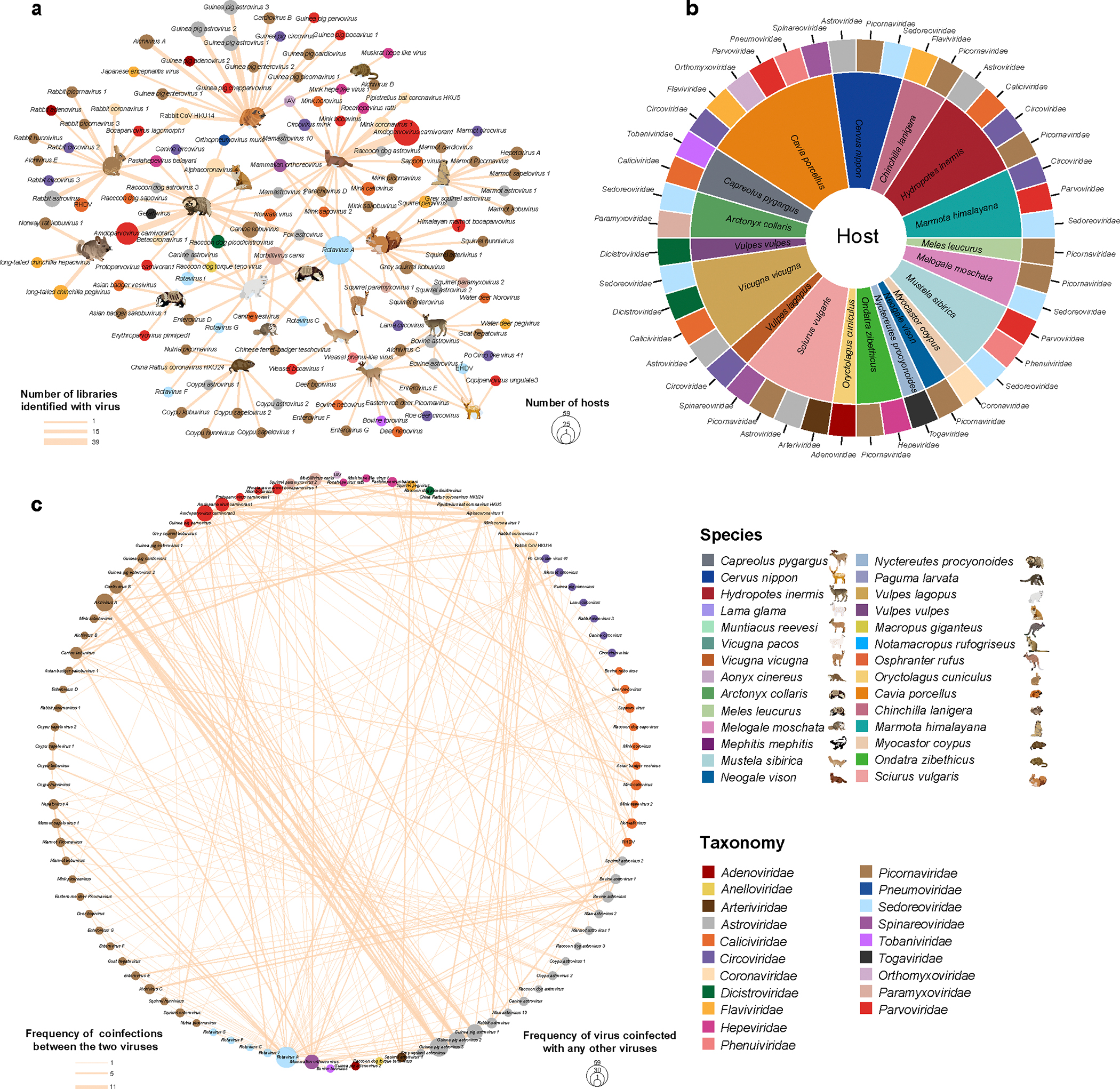
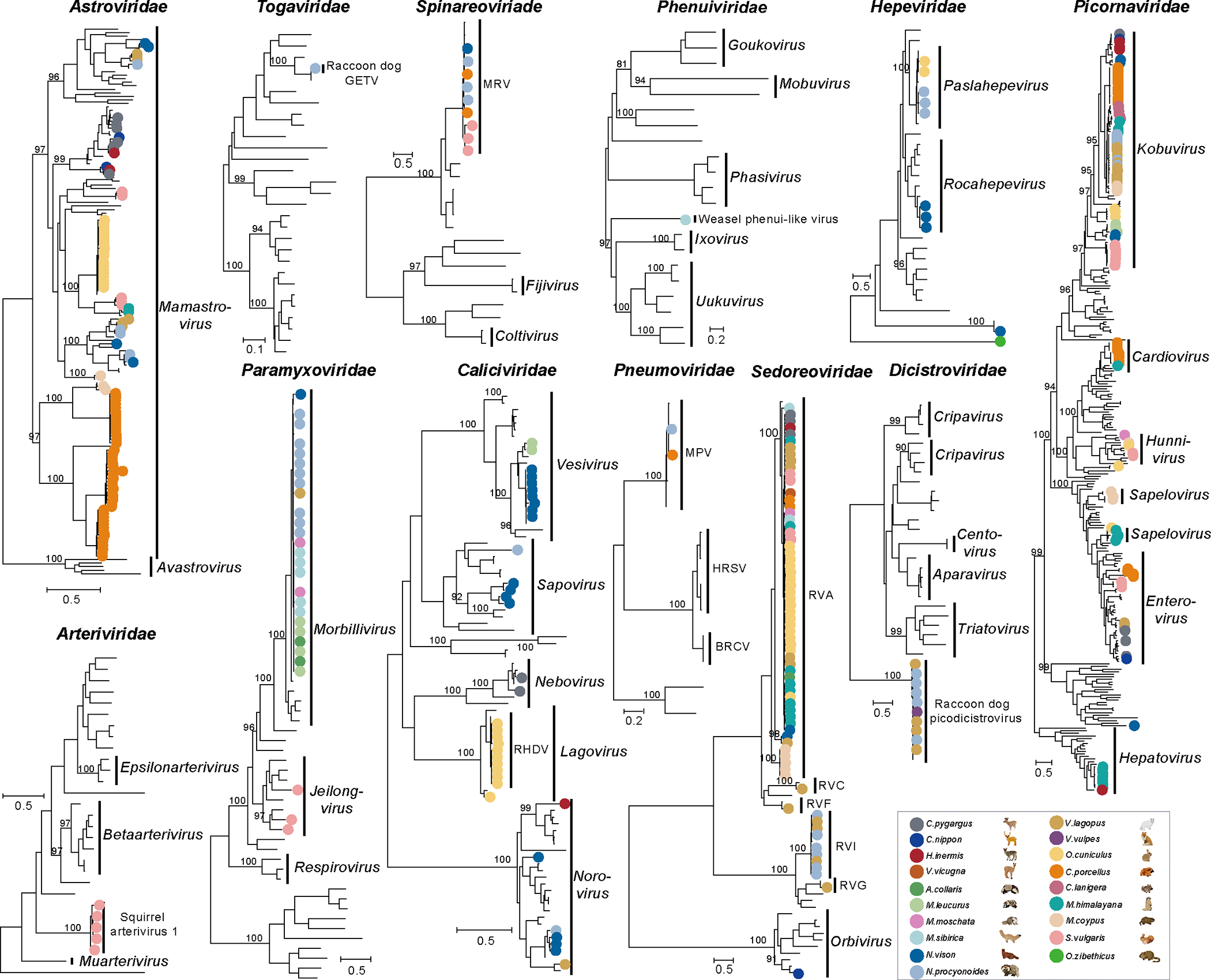
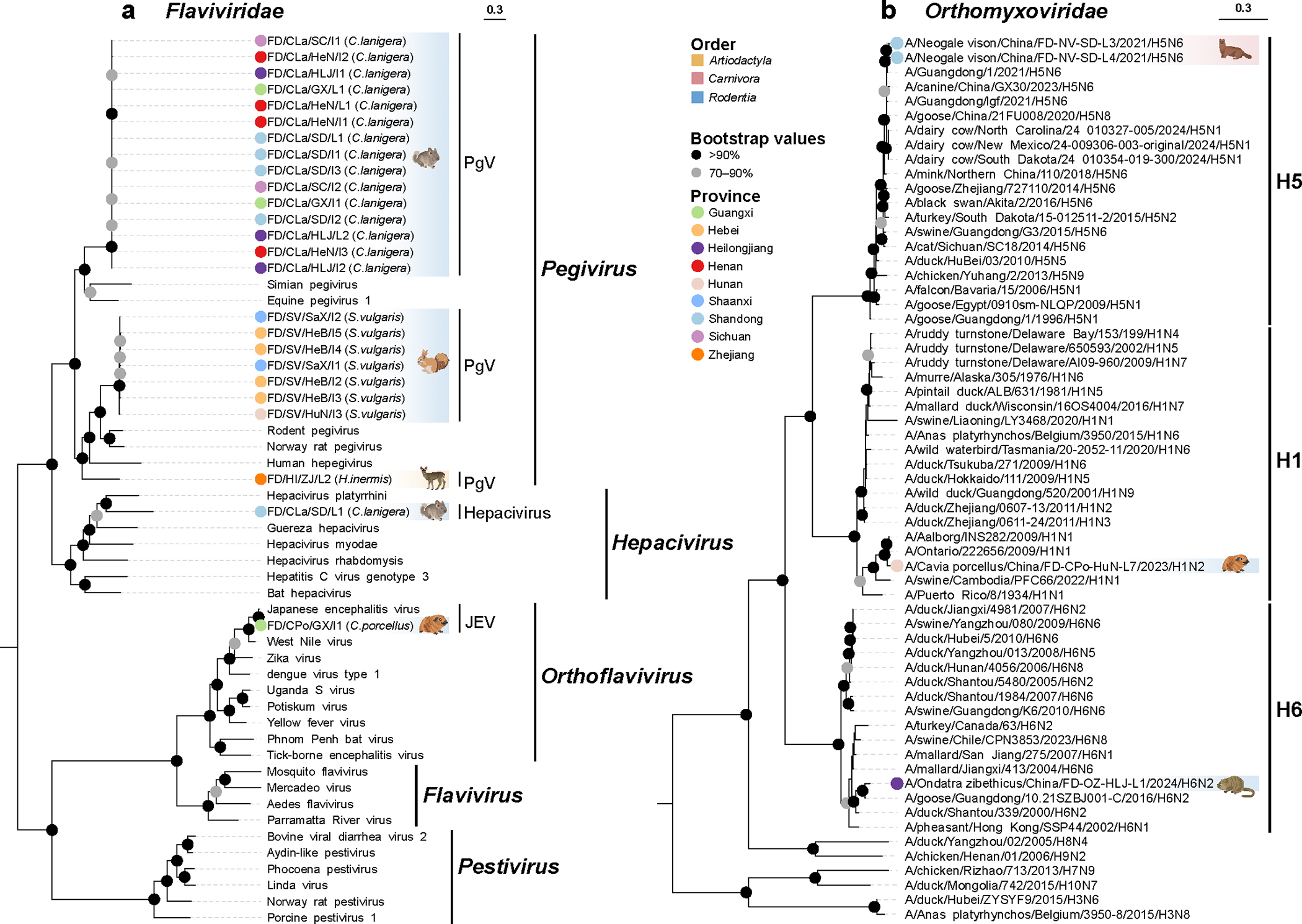

References
-
- Lindh E et al. Highly pathogenic avian influenza A(H5N1) virus infection on multiple fur farms in the South and Central Ostrobothnia regions of Finland, July 2023. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin 28, doi: 10.2807/1560-7917.Es.2023.28.31.2300400 (2023). - DOI - PMC - PubMed
