

Chromosome 16p11.2 microdeletion syndrome with microcephaly and Dandy-Walker malformation spectrum: expanding the known phenotype
- PMID: 39232803
- PMCID: PMC11376027
- DOI: 10.1186/s40246-024-00662-0
Chromosome 16p11.2 microdeletion syndrome with microcephaly and Dandy-Walker malformation spectrum: expanding the known phenotype
Erratum in
-
Correction: Chromosome 16p11.2 microdeletion syndrome with microcephaly and Dandy-Walker malformation spectrum: expanding the known phenotype.Hum Genomics. 2024 Oct 15;18(1):115. doi: 10.1186/s40246-024-00681-x. Hum Genomics. 2024. PMID: 39407347 Free PMC article. No abstract available.
Abstract
Background: Chromosome 16p11.2 deletions and duplications were found to be the second most common copy number variation (CNV) reported in cases with clinical presentation suggestive of chromosomal syndromes. Chromosome 16p11.2 deletion syndrome shows remarkable phenotypic heterogeneity with a wide variability of presentation extending from normal development and cognition to severe phenotypes. The clinical spectrum ranges from neurocognitive and global developmental delay (GDD), intellectual disability, and language defects (dysarthria /apraxia) to neuropsychiatric and autism spectrum disorders. Other presentations include dysmorphic features, congenital malformations, insulin resistance, and a tendency for obesity. Our study aims to narrow the gap of knowledge in Saudi Arabia and the Middle Eastern and Northern African (MENA) region about genetic disorders, particularly CNV-associated disorders. Despite their rarity, genetic studies in the MENA region revealed high potential with remarkable genetic and phenotypic novelty.
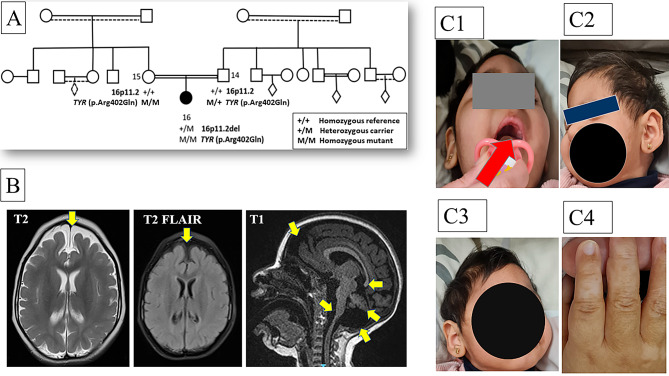
Results: We identified a heterozygous de novo recurrent proximal chromosome 16p11.2 microdeletion by microarray (arr[GRCh38]16p11.2(29555974_30166595)x1) [(arr[GRCh37]16p11.2(29567295_30177916)x1)] and confirmed by whole exome sequencing (arr[GRCh37]16p11.2(29635211_30199850)x1). We report a Saudi girl with severe motor and cognitive disability, myoclonic epilepsy, deafness, and visual impairment carrying the above-described deletion. Our study broadens the known phenotypic spectrum associated with recurrent proximal 16p11.2 microdeletion syndrome to include developmental dysplasia of the hip, optic atrophy, and a flat retina. Notably, the patient exhibited a rare combination of microcephaly, features consistent with the Dandy-Walker spectrum, and a thin corpus callosum (TCC), which are extremely infrequent presentations in patients with the 16p11.2 microdeletion. Additionally, the patient displayed areas of skin and hair hypopigmentation, attributed to a homozygous hypomorphic allele in the TYR gene.
Conclusion: This report expands on the clinical phenotype associated with proximal 16p11.2 microdeletion syndrome, highlighting the potential of genetic research in Saudi Arabia and the MENA region. It underscores the importance of similar future studies.
Keywords: Cognitive impairment; Dandy-walker spectrum; Global developmental delay; Microcephaly; Myoclonic epilepsy; Optic atrophy; Recurrent proximal chromosome 16p11.2 microdeletion syndrome; Saudi Arabia; de novo.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Oliva-Teles N, de Stefano MC, Gallagher L, Rakic S, Jorge P, Cuturilo G, et al. Rare pathogenic Copy Number Variation in the 16p11.2 (BP4-BP5) Region Associated with Neurodevelopmental and Neuropsychiatric disorders: a review of the literature. Int J Environ Res Public Health. 2020;17:9253. - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
