

Engineering Pyrrolysine Systems for Genetic Code Expansion and Reprogramming
- PMID: 39235427
- PMCID: PMC11467909
- DOI: 10.1021/acs.chemrev.4c00243
Engineering Pyrrolysine Systems for Genetic Code Expansion and Reprogramming
Abstract
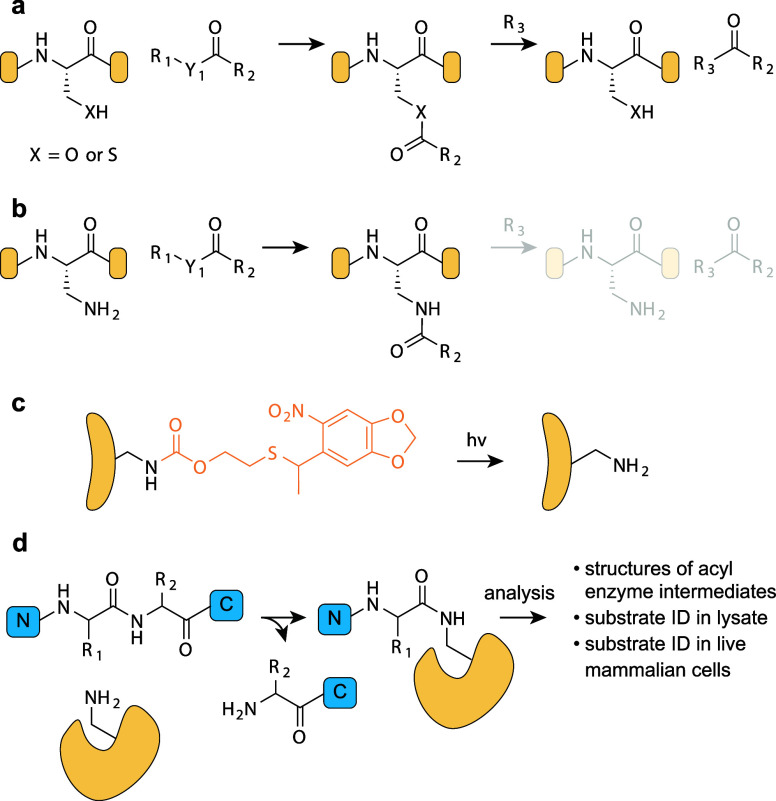
Over the past 16 years, genetic code expansion and reprogramming in living organisms has been transformed by advances that leverage the unique properties of pyrrolysyl-tRNA synthetase (PylRS)/tRNAPyl pairs. Here we summarize the discovery of the pyrrolysine system and describe the unique properties of PylRS/tRNAPyl pairs that provide a foundation for their transformational role in genetic code expansion and reprogramming. We describe the development of genetic code expansion, from E. coli to all domains of life, using PylRS/tRNAPyl pairs, and the development of systems that biosynthesize and incorporate ncAAs using pyl systems. We review applications that have been uniquely enabled by the development of PylRS/tRNAPyl pairs for incorporating new noncanonical amino acids (ncAAs), and strategies for engineering PylRS/tRNAPyl pairs to add noncanonical monomers, beyond α-L-amino acids, to the genetic code of living organisms. We review rapid progress in the discovery and scalable generation of mutually orthogonal PylRS/tRNAPyl pairs that can be directed to incorporate diverse ncAAs in response to diverse codons, and we review strategies for incorporating multiple distinct ncAAs into proteins using mutually orthogonal PylRS/tRNAPyl pairs. Finally, we review recent advances in the encoded cellular synthesis of noncanonical polymers and macrocycles and discuss future developments for PylRS/tRNAPyl pairs.
Conflict of interest statement
The authors declare the following competing financial interest(s): J.W.C. is a founder of Constructive Bio. D.L.D is a consultant for Constructive Bio.
Figures































References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
