

Oncolytic virotherapy augments self-maintaining natural killer cell line cytotoxicity against neuroblastoma
- PMID: 39235531
- PMCID: PMC11377387
- DOI: 10.1007/s00262-024-03818-y
Oncolytic virotherapy augments self-maintaining natural killer cell line cytotoxicity against neuroblastoma
Abstract
Background: Neuroblastoma is the most common extracranial solid tumor in children and accounts for 15% of pediatric cancer related deaths. Targeting neuroblastoma with immunotherapies has proven challenging due to a paucity of immune cells in the tumor microenvironment and the release of immunosuppressive cytokines by neuroblastoma tumor cells. We hypothesized that combining an oncolytic Herpes Simplex Virus (oHSV) with natural killer (NK) cells might overcome these barriers and incite tumor cell death.
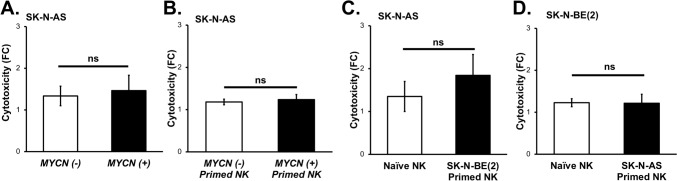
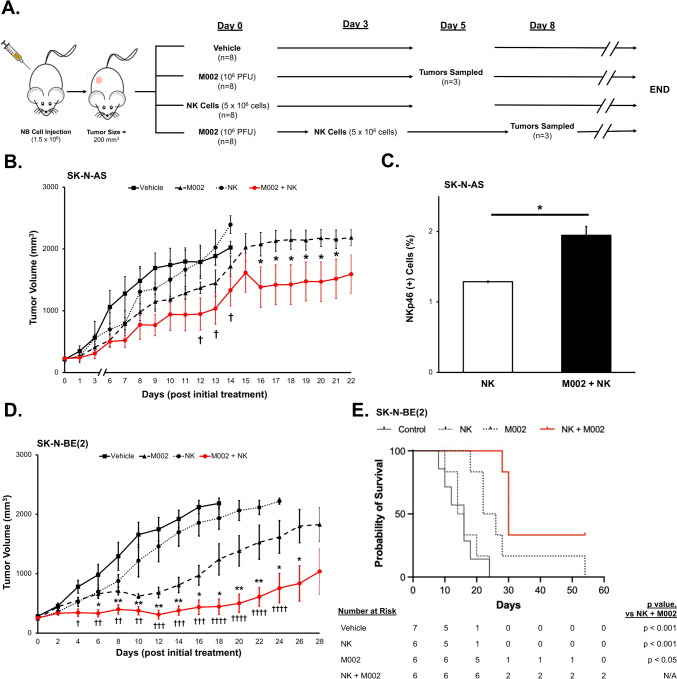
Methods: We utilized MYCN amplified and non-amplified neuroblastoma cell lines, the IL-12 expressing oHSV, M002, and the human NK cell line, NK-92 MI. We assessed the cytotoxicity of NK cells against neuroblastoma with and without M002 infection, the effects of M002 on NK cell priming, and the impact of M002 and priming on the migratory capacity and CD107a expression of NK cells. To test clinical applicability, we then investigated the effects of M002 and NK cells on neuroblastoma in vivo.
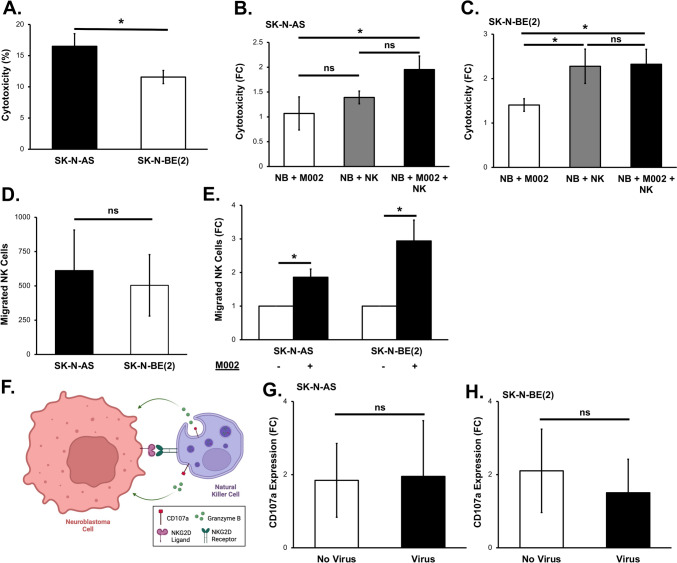
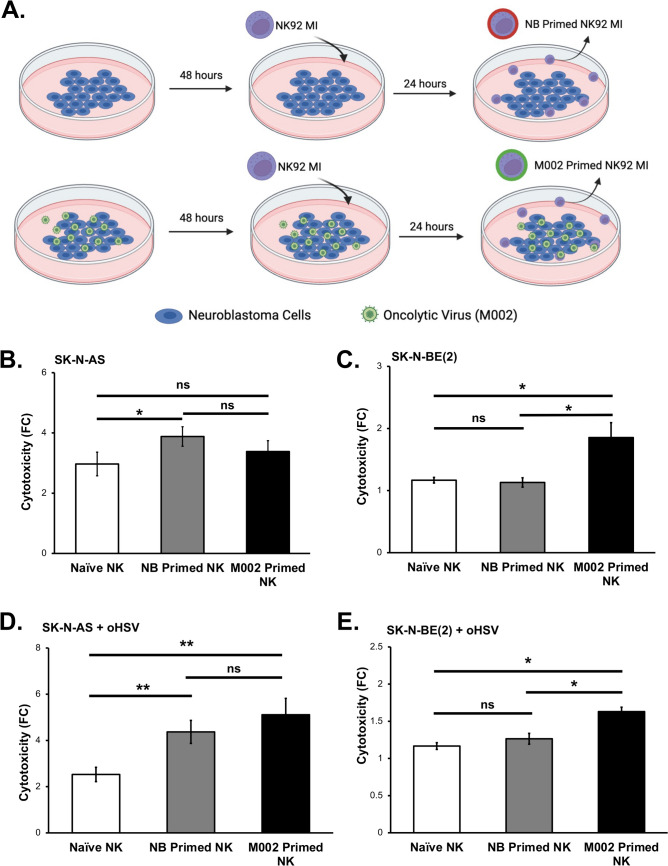
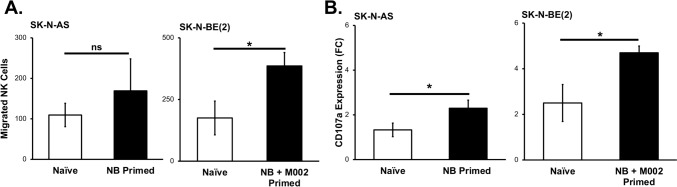
Results: NK cells were more attracted to neuroblastoma cells that were infected with M002. There was an increase in neuroblastoma cell death with the combination treatment of M002 and NK cells both in vitro and in vivo. Priming the NK cells enhanced their cytotoxicity, migratory capacity and CD107a expression.
Conclusions: To the best of our knowledge, these investigations are the first to demonstrate the effects of an oncolytic virus combined with self-maintaining NK cells in neuroblastoma and the priming effect of neuroblastoma on NK cells. The current studies provide a deeper understanding of the relation between NK cells and neuroblastoma and these data suggest that oHSV increases NK cell cytotoxicity towards neuroblastoma.
Keywords: Immunotherapy; Natural killer cells; Neuroblastoma; Oncolytic virus; Translational research.
© 2024. The Author(s).
Conflict of interest statement
Dr J.M. Markert holds equity in Aettis, Inc. (< 8%), a company that holds stocks of oncolytic virus and Treovir, Inc. (25%), a company holding intellectual property and funding clinical trials of oncolytic virus for pediatric brain tumors. A company that Dr J.M. Markert formerly held equity in (< 8%) Catherex, Inc., was purchased in a structured buyout. Dr J.M. Markert has served as a consultant for Imugene. He also holds a fraction of the IP associated with oncolytic virus C134, which is licensed by Mustang Biotech.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
