

Obtention of viable cell suspensions from breast cancer tumor biopsies for 3D chromatin conformation and single-cell transcriptome analysis
- PMID: 39239354
- PMCID: PMC11375512
- DOI: 10.3389/fmolb.2024.1420308
Obtention of viable cell suspensions from breast cancer tumor biopsies for 3D chromatin conformation and single-cell transcriptome analysis
Abstract
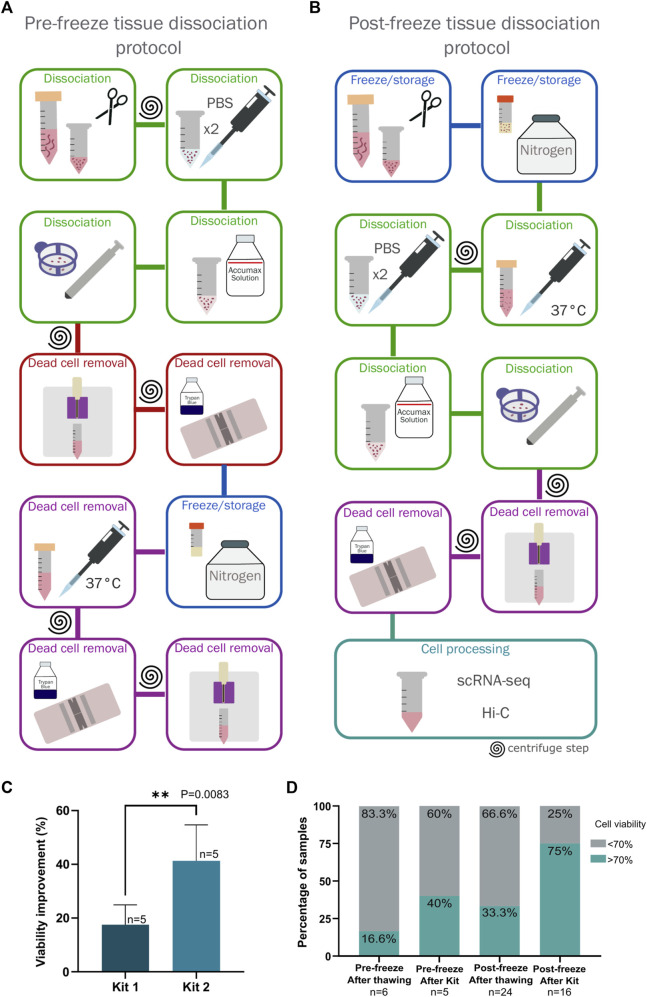
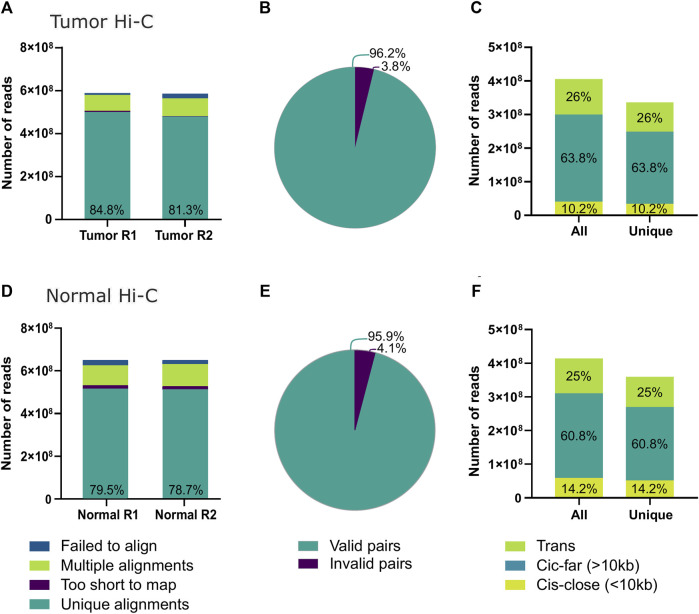
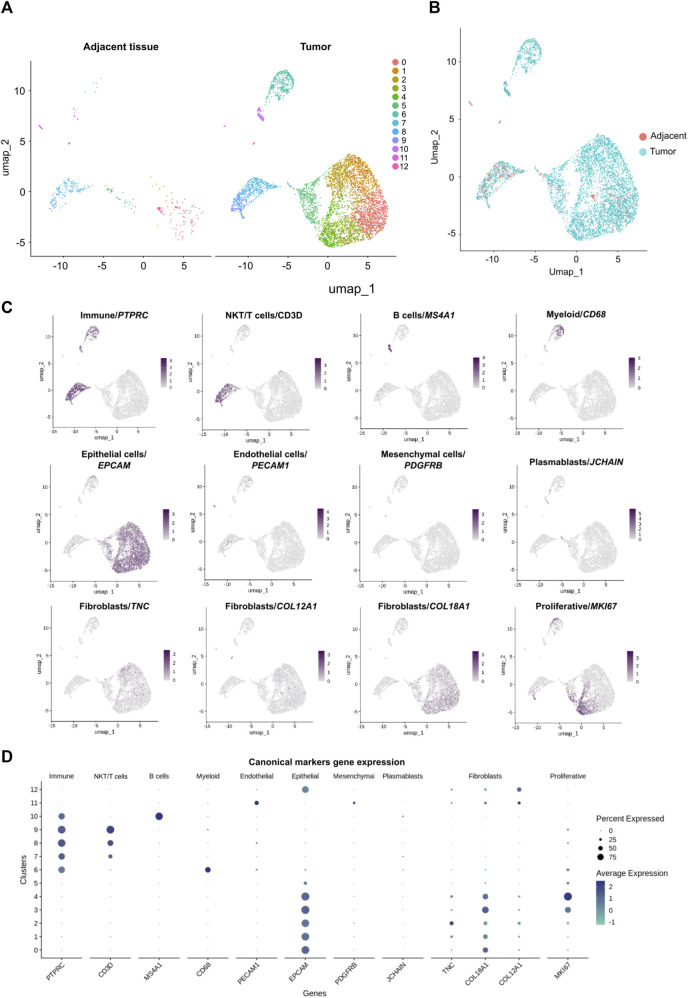
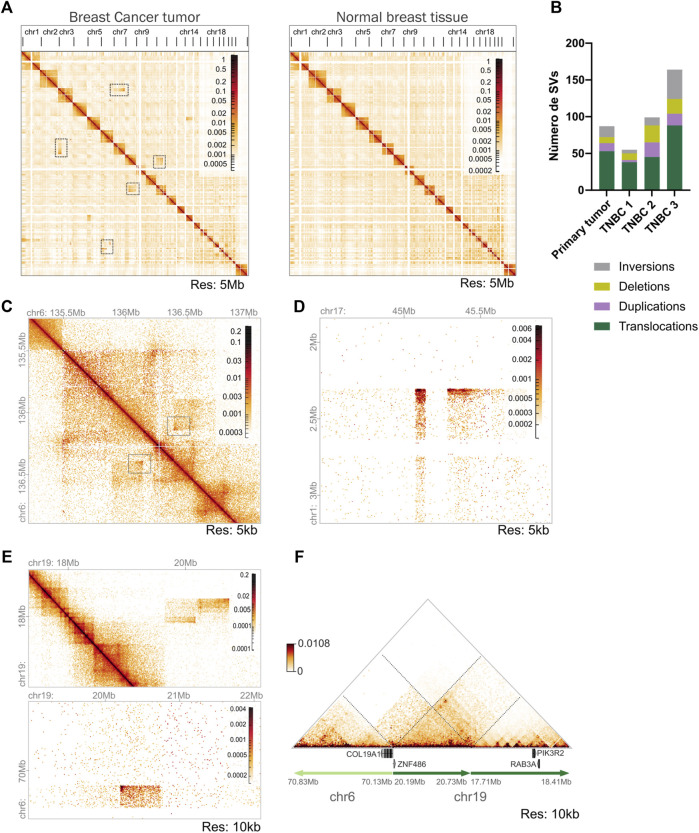
Molecular and cellular characterization of tumors is essential due to the complex and heterogeneous nature of cancer. In recent decades, many bioinformatic tools and experimental techniques have been developed to achieve personalized characterization of tumors. However, sample handling continues to be a major challenge as limitations such as prior treatments before sample acquisition, the amount of tissue obtained, transportation, or the inability to process fresh samples pose a hurdle for experimental strategies that require viable cell suspensions. Here, we present an optimized protocol that allows the recovery of highly viable cell suspensions from breast cancer primary tumor biopsies. Using these cell suspensions we have successfully characterized genome architecture through Hi-C. Also, we have evaluated single-cell gene expression and the tumor cellular microenvironment through single-cell RNAseq. Both technologies are key in the detailed and personalized molecular characterization of tumor samples. The protocol described here is a cost-effective alternative to obtain viable cell suspensions from biopsies simply and efficiently.
Keywords: 3D genome architecture; Hi-C; breast cancer; single cell RNA sequencing; structural variations (SVs).
Copyright © 2024 Stephenson-Gussinye, Rendón-Bautista, Ruiz-Medina, Blanco-Olais, Pérez-Molina, Marcial-Medina, Chavarri-Guerra, Soto-Pérez-de-Celis, Morales-Alfaro, Esquivel-López, Candanedo-González, Gamboa-Domínguez, Cortes-González, Alfaro-Goldaracena, Vázquez-Manjarrez, Grajales-Figueroa, Astudillo-Romero, Ruiz-Manriquez, Poot-Hernández, Licona-Limón and Furlan-Magaril.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
Similar articles
-
3D disorganization and rearrangement of genome provide insights into pathogenesis of NAFLD by integrated Hi-C, Nanopore, and RNA sequencing.Acta Pharm Sin B. 2021 Oct;11(10):3150-3164. doi: 10.1016/j.apsb.2021.03.022. Epub 2021 Apr 6. Acta Pharm Sin B. 2021. PMID: 34729306 Free PMC article.
-
Dissociation of Human and Mouse Tumor Tissue Samples for Single-cell RNA Sequencing.J Vis Exp. 2024 Aug 16;(210). doi: 10.3791/66766. J Vis Exp. 2024. PMID: 39221955
-
Three-dimensional genome landscape comprehensively reveals patterns of spatial gene regulation in papillary and anaplastic thyroid cancers: a study using representative cell lines for each cancer type.Cell Mol Biol Lett. 2023 Jan 6;28(1):1. doi: 10.1186/s11658-022-00409-6. Cell Mol Biol Lett. 2023. PMID: 36609218 Free PMC article.
-
Hi-C as a molecular rangefinder to examine genomic rearrangements.Semin Cell Dev Biol. 2022 Jan;121:161-170. doi: 10.1016/j.semcdb.2021.04.024. Epub 2021 May 13. Semin Cell Dev Biol. 2022. PMID: 33992531 Review.
-
[Mass spectrometry imaging technology and its application in breast cancer research].Se Pu. 2021 Jun;39(6):578-587. doi: 10.3724/SP.J.1123.2020.10005. Se Pu. 2021. PMID: 34227318 Free PMC article. Review. Chinese.
References
-
- Cancemi P., Buttacavoli M., Roz E., Feo S. (2019). Expression of alpha-enolase (ENO1), myc promoter-binding protein-1 (MBP-1) and matrix metalloproteinases (MMP-2 and MMP-9) reflect the nature and aggressiveness of breast tumors. Int. J. Mol. Sci. 20 (16), 3952. 10.3390/ijms20163952 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases