

Acutely blocking excessive mitochondrial fission prevents chronic neurodegeneration after traumatic brain injury
- PMID: 39241772
- PMCID: PMC11525032
- DOI: 10.1016/j.xcrm.2024.101715
Acutely blocking excessive mitochondrial fission prevents chronic neurodegeneration after traumatic brain injury
Abstract
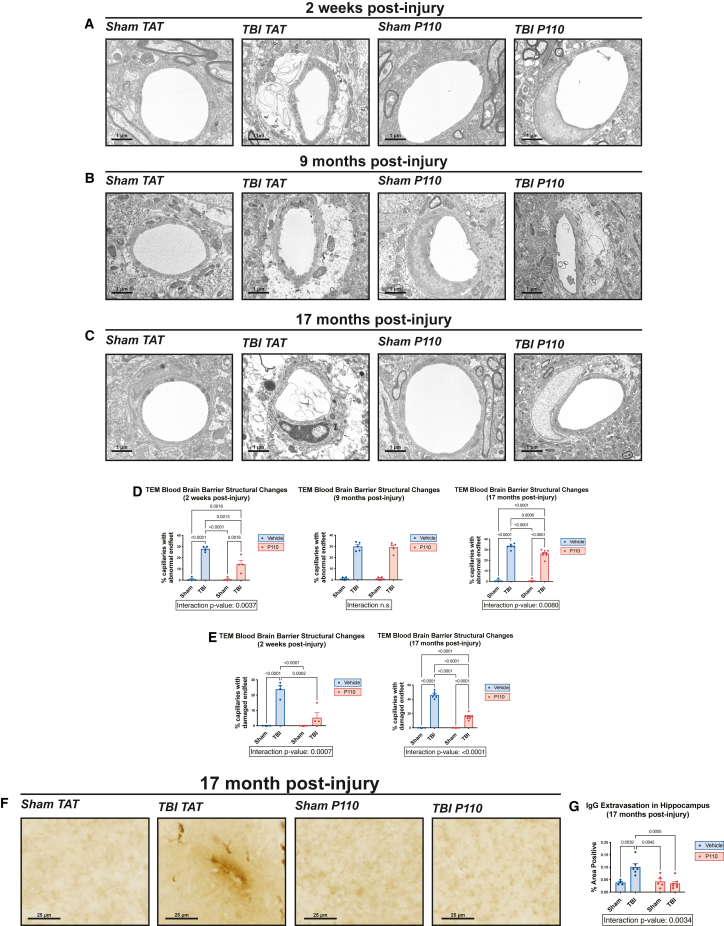
Progression of acute traumatic brain injury (TBI) into chronic neurodegeneration is a major health problem with no protective treatments. Here, we report that acutely elevated mitochondrial fission after TBI in mice triggers chronic neurodegeneration persisting 17 months later, equivalent to many human decades. We show that increased mitochondrial fission after mouse TBI is related to increased brain levels of mitochondrial fission 1 protein (Fis1) and that brain Fis1 is also elevated in human TBI. Pharmacologically preventing Fis1 from binding its mitochondrial partner, dynamin-related protein 1 (Drp1), for 2 weeks after TBI normalizes the balance of mitochondrial fission/fusion and prevents chronically impaired mitochondrial bioenergetics, oxidative damage, microglial activation and lipid droplet formation, blood-brain barrier deterioration, neurodegeneration, and cognitive impairment. Delaying treatment until 8 months after TBI offers no protection. Thus, time-sensitive inhibition of acutely elevated mitochondrial fission may represent a strategy to protect human TBI patients from chronic neurodegeneration.
Keywords: Drp1; Fis1; blood-brain barrier; mitochondria; mitochondrial fission; mitochondrial fusion; neurodegeneration; neuroprotection; oxidative stress; traumatic brain injury.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests X.Q. is an inventor of P110 and holds patents related to P110.
Figures







References
-
- Ma V.Y., Chan L., Carruthers K.J. Incidence, Prevalence, Costs, and Impact on Disability of Common Conditions Requiring Rehabilitation in the United States: Stroke, Spinal Cord Injury, Traumatic Brain Injury, Multiple Sclerosis, Osteoarthritis, Rheumatoid Arthritis, Limb Loss, and Back Pain. Arch. Phys. Med. Rehabil. 2014;95:986–995.e1. doi: 10.1016/j.apmr.2013.10.032. - DOI - PMC - PubMed
-
- McKee A.C., Cairns N.J., Dickson D.W., Folkerth R.D., Keene C.D., Litvan I., Perl D.P., Stein T.D., Vonsattel J.-P., Stewart W., et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131:75–86. doi: 10.1007/s00401-015-1515-z. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- T32 GM152319/GM/NIGMS NIH HHS/United States
- R01 AG071512/AG/NIA NIH HHS/United States
- R01 AG076051/AG/NIA NIH HHS/United States
- RF1 AG074346/AG/NIA NIH HHS/United States
- R01 AG066707/AG/NIA NIH HHS/United States
- R01 AG065240/AG/NIA NIH HHS/United States
- T32 AG071474/AG/NIA NIH HHS/United States
- P30 AG072977/AG/NIA NIH HHS/United States
- R01 AG059721/AG/NIA NIH HHS/United States
- T32 GM007250/GM/NIGMS NIH HHS/United States
- I01 BX005976/BX/BLRD VA/United States
- R01 NS115903/NS/NINDS NIH HHS/United States
- RF1 NS122218/NS/NINDS NIH HHS/United States
- R01 AG067741/AG/NIA NIH HHS/United States
- R21 AG073684/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
