

Intracellular spatial transcriptomic analysis toolkit (InSTAnT)
- PMID: 39242579
- PMCID: PMC11379969
- DOI: 10.1038/s41467-024-49457-w
Intracellular spatial transcriptomic analysis toolkit (InSTAnT)
Erratum in
-
Author Correction: Intracellular spatial transcriptomic analysis toolkit (InSTAnT).Nat Commun. 2024 Oct 25;15(1):9219. doi: 10.1038/s41467-024-53244-y. Nat Commun. 2024. PMID: 39455563 Free PMC article. No abstract available.
Abstract
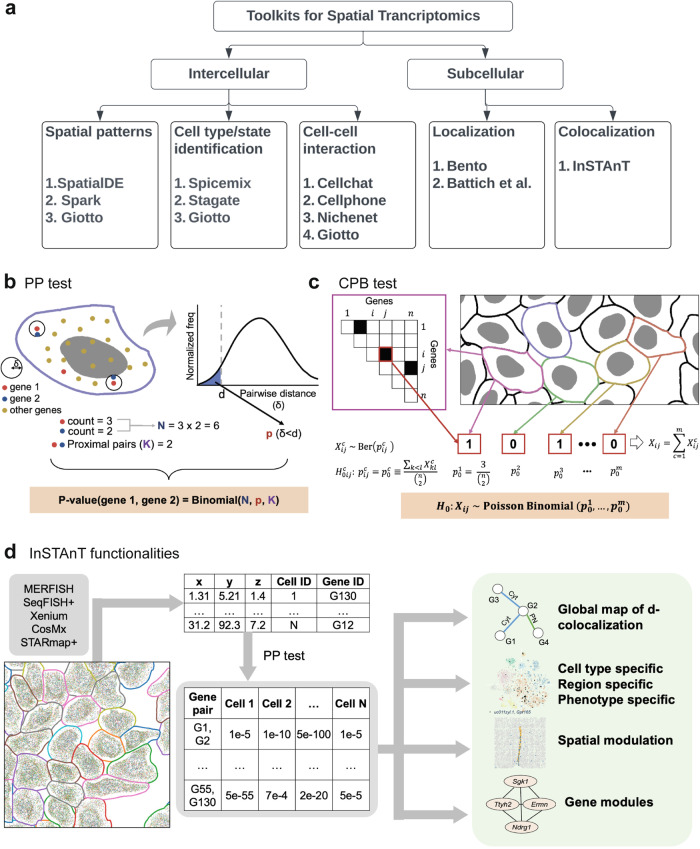
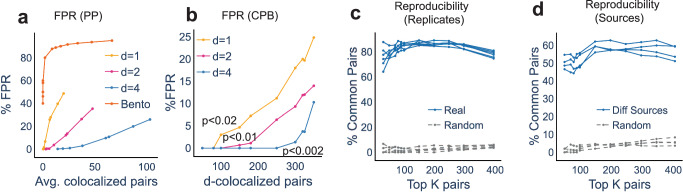
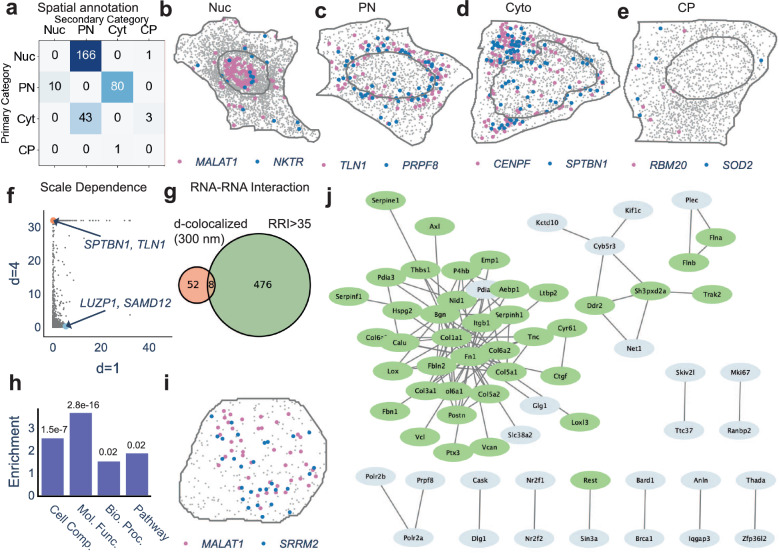
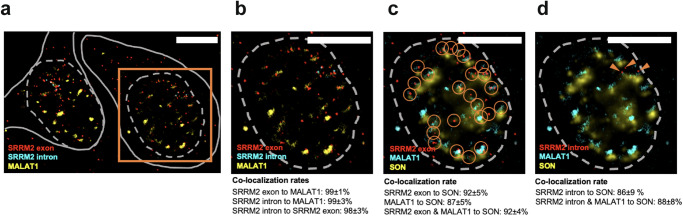
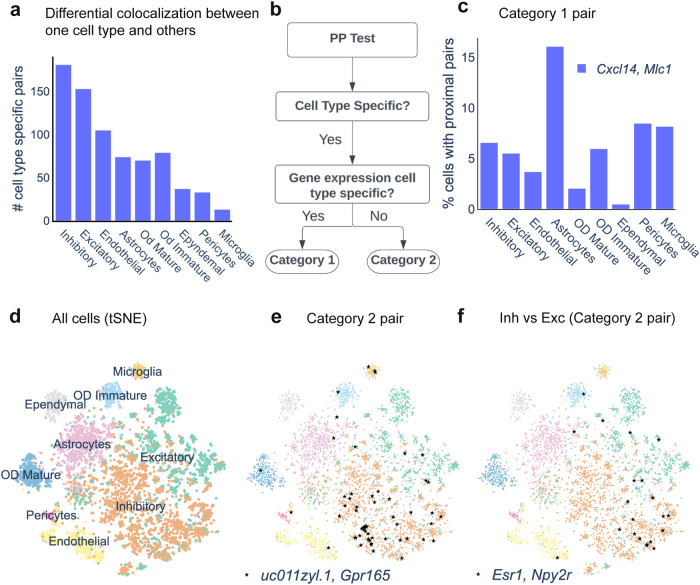
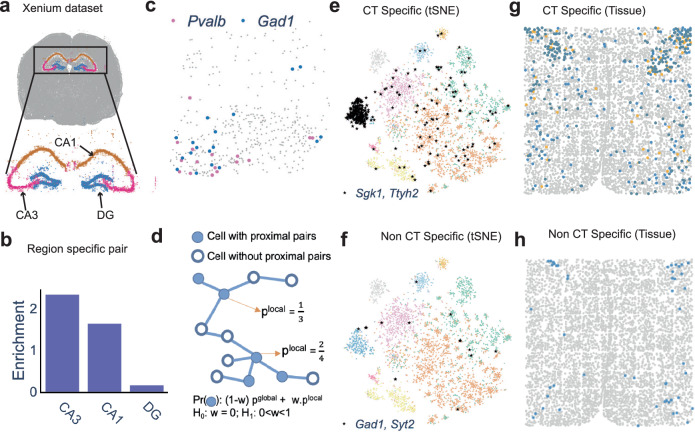
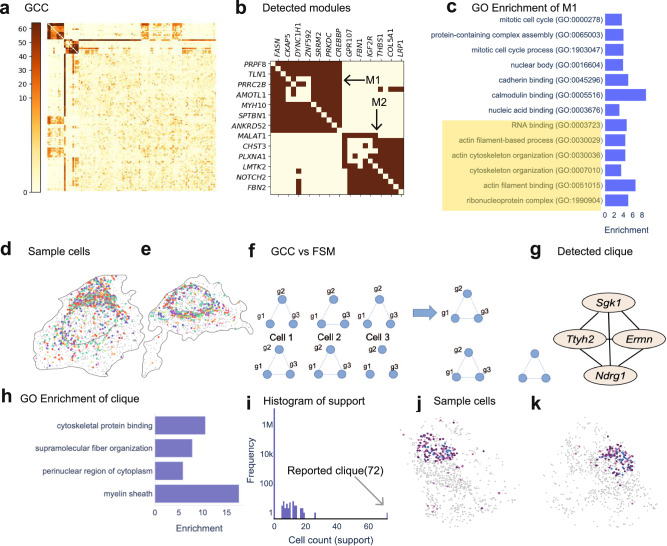
Imaging-based spatial transcriptomics technologies such as Multiplexed error-robust fluorescence in situ hybridization (MERFISH) can capture cellular processes in unparalleled detail. However, rigorous and robust analytical tools are needed to unlock their full potential for discovering subcellular biological patterns. We present Intracellular Spatial Transcriptomic Analysis Toolkit (InSTAnT), a computational toolkit for extracting molecular relationships from spatial transcriptomics data at single molecule resolution. InSTAnT employs specialized statistical tests and algorithms to detect gene pairs and modules exhibiting intriguing patterns of co-localization, both within individual cells and across the cellular landscape. We showcase the toolkit on five different datasets representing two different cell lines, two brain structures, two species, and three different technologies. We perform rigorous statistical assessment of discovered co-localization patterns, find supporting evidence from databases and RNA interactions, and identify associated subcellular domains. We uncover several cell type and region-specific gene co-localizations within the brain. Intra-cellular spatial patterns discovered by InSTAnT mirror diverse molecular relationships, including RNA interactions and shared sub-cellular localization or function, providing a rich compendium of testable hypotheses regarding molecular functions.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Intracellular Spatial Transcriptomic Analysis Toolkit (InSTAnT).Res Sq [Preprint]. 2023 Jan 27:rs.3.rs-2481749. doi: 10.21203/rs.3.rs-2481749/v1. Res Sq. 2023. Update in: Nat Commun. 2024 Sep 6;15(1):7794. doi: 10.1038/s41467-024-49457-w. PMID: 36747718 Free PMC article. Updated. Preprint.
References
-
- Marx, V. Method of the Year: spatially resolved transcriptomics. Nat. methods18, 9–14 (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R35 GM131819/GM/NIGMS NIH HHS/United States
- R35 GM147420/GM/NIGMS NIH HHS/United States
- T32 GM136629/GM/NIGMS NIH HHS/United States
- R35GM131819/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R21HG013180/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
