

Population genomics of Streptococcus mitis in UK and Ireland bloodstream infection and infective endocarditis cases
- PMID: 39242612
- PMCID: PMC11379897
- DOI: 10.1038/s41467-024-52120-z
Population genomics of Streptococcus mitis in UK and Ireland bloodstream infection and infective endocarditis cases
Abstract
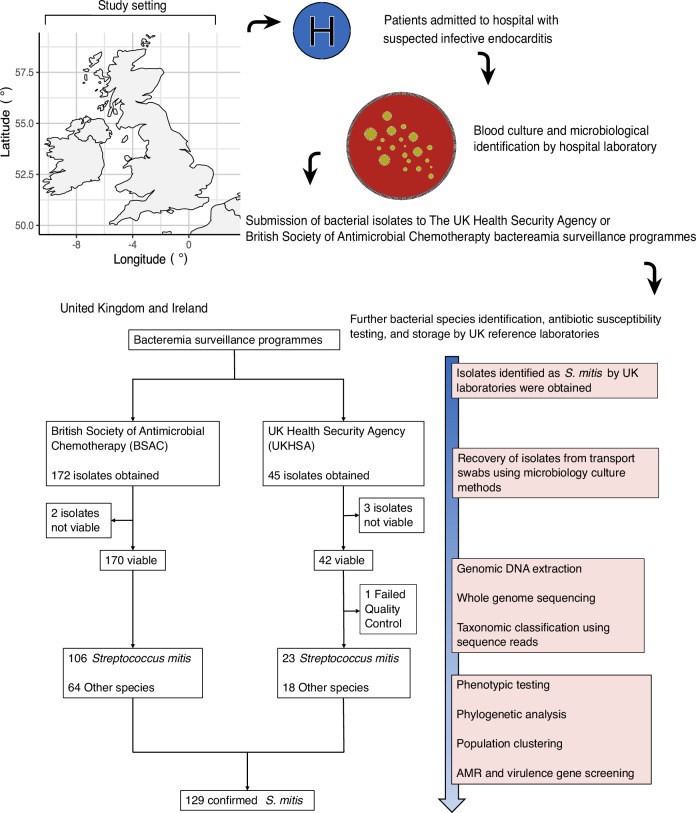
Streptococcus mitis is a leading cause of infective endocarditis (IE). However, our understanding of the genomic epidemiology and pathogenicity of IE-associated S. mitis is hampered by low IE incidence. Here we use whole genome sequencing of 129 S. mitis bloodstream infection (BSI) isolates collected between 2001-2016 from clinically diagnosed IE cases in the UK to investigate genetic diversity, antimicrobial resistance, and pathogenicity. We show high genetic diversity of IE-associated S. mitis with virtually all isolates belonging to distinct lineages indicating no predominance of specific lineages. Additionally, we find a highly variable distribution of known pneumococcal virulence genes among the isolates, some of which are overrepresented in disease when compared to carriage strains. Our findings suggest that S. mitis in patients with clinically diagnosed IE is not primarily caused by specific hypervirulent or antimicrobial resistant lineages, highlighting the accidental pathogenic nature of S. mitis in patients with clinically diagnosed IE.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
