

Metagenomic analysis reveals high diversity of gut viromes in yaks (Bos grunniens) from the Qinghai-Tibet Plateau
- PMID: 39242698
- PMCID: PMC11379701
- DOI: 10.1038/s42003-024-06798-y
Metagenomic analysis reveals high diversity of gut viromes in yaks (Bos grunniens) from the Qinghai-Tibet Plateau
Abstract
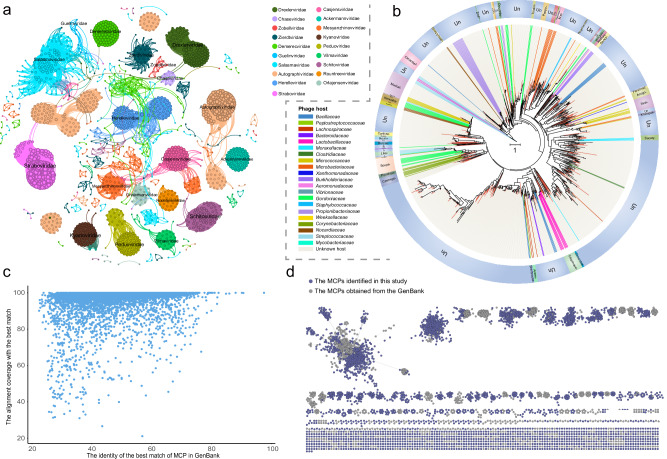
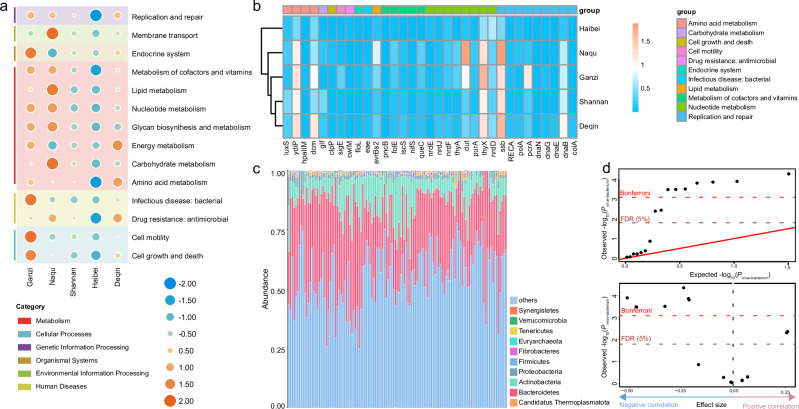
The Qinghai-Tibet Plateau (QTP), renowned for its exceptional biological diversity, is home to numerous endemic species. However, research on the virology of vulnerable vertebrates like yaks remains limited. In this study, our objective was to use metagenomics to provide a comprehensive understanding of the diversity and evolution of the gut virome in yak populations across different regions of the QTP. Our findings revealed a remarkably diverse array of viruses in the gut of yaks, including those associated with vertebrates and bacteriophages. Notably, some vertebrate-associated viruses, such as astrovirus and picornavirus, showed significant sequence identity across diverse yak populations. Additionally, we observed differences in the functional profiles of genes carried by the yak gut virome across different regions. Moreover, the virus-bacterium symbiotic network that we discovered holds potential significance in maintaining the health of yaks. Overall, this research expands our understanding of the viral communities in the gut of yaks and highlights the importance of further investigating the interactions between viruses and their hosts. These data will be beneficial for revealing the crucial role that viruses play in the yak gut ecology in future studies.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
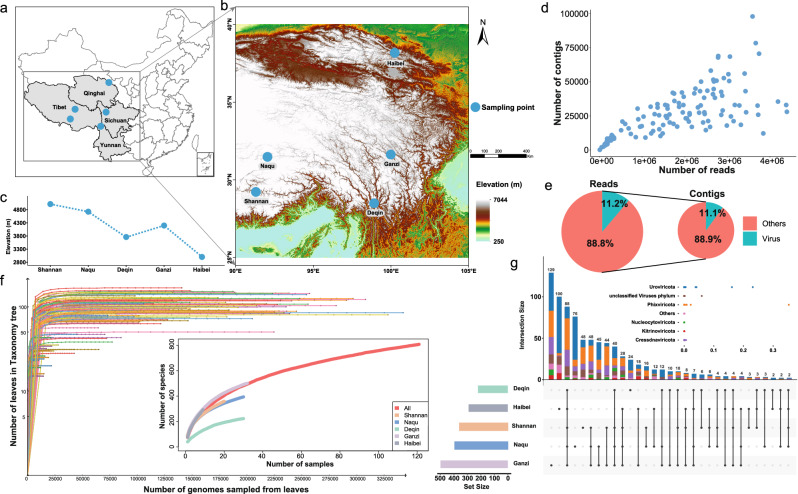
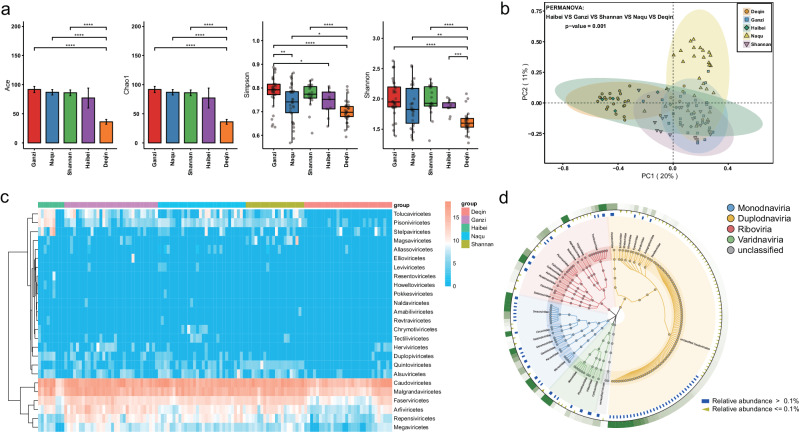
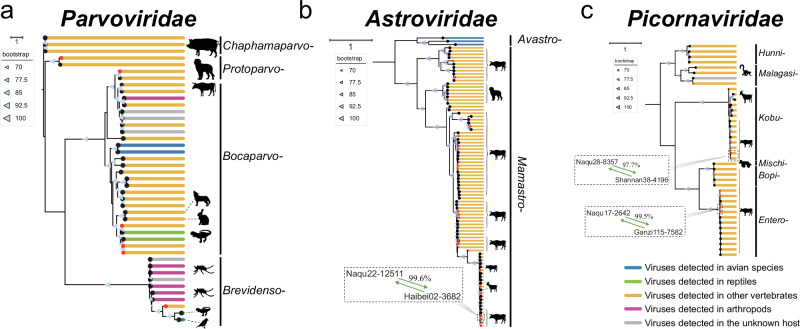
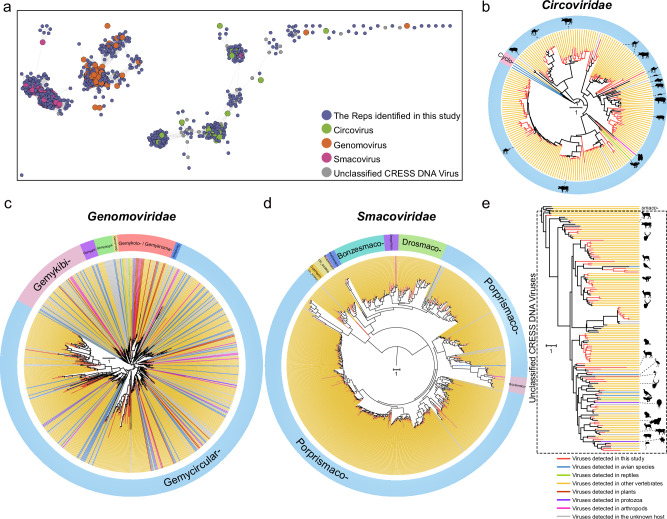
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
