

Primary succession of Bifidobacteria drives pathogen resistance in neonatal microbiota assembly
- PMID: 39242817
- PMCID: PMC11445081
- DOI: 10.1038/s41564-024-01804-9
Primary succession of Bifidobacteria drives pathogen resistance in neonatal microbiota assembly
Abstract
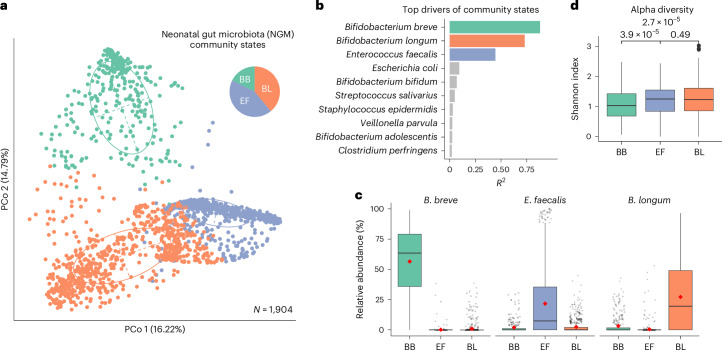
Human microbiota assembly commences at birth, seeded by both maternal and environmental microorganisms. Ecological theory postulates that primary colonizers dictate microbial community assembly outcomes, yet such microbial priority effects in the human gut remain underexplored. Here using longitudinal faecal metagenomics, we characterized neonatal microbiota assembly for a cohort of 1,288 neonates from the UK. We show that the pioneering neonatal gut microbiota can be stratified into one of three distinct community states, each dominated by a single microbial species and influenced by clinical and host factors, such as maternal age, ethnicity and parity. A community state dominated by Enterococcus faecalis displayed stochastic microbiota assembly with persistent high pathogen loads into infancy. In contrast, community states dominated by Bifidobacterium, specifically B. longum and particularly B. breve, exhibited a stable assembly trajectory and long-term pathogen colonization resistance, probably due to strain-specific functional adaptions to a breast milk-rich neonatal diet. Consistent with our human cohort observation, B. breve demonstrated priority effects and conferred pathogen colonization resistance in a germ-free mouse model. Our findings solidify the crucial role of Bifidobacteria as primary colonizers in shaping the microbiota assembly and functions in early life.
© 2024. The Author(s).
Conflict of interest statement
T.D.L. is the co-founder and CSO of Microbiotica. The other authors declare no competing interests.
Figures












References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
