

Molecular epidemiology and evolutionary characteristics of dengue virus 2 in East Africa
- PMID: 39244569
- PMCID: PMC11380673
- DOI: 10.1038/s41467-024-51018-0
Molecular epidemiology and evolutionary characteristics of dengue virus 2 in East Africa
Abstract
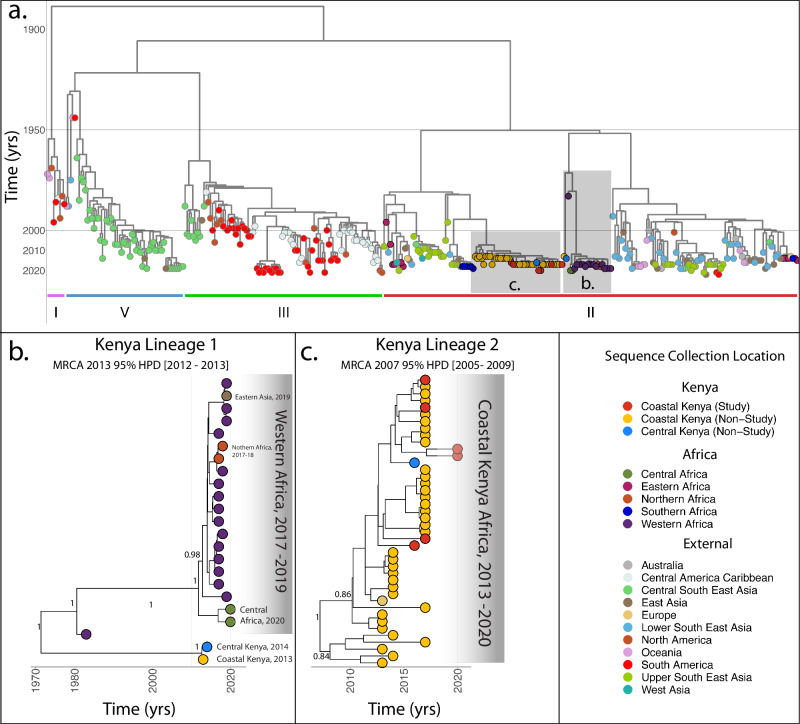
Despite the increasing burden of dengue, the regional emergence of the virus in Kenya has not been examined. This study investigates the genetic structure and regional spread of dengue virus-2 in Kenya. Viral RNA from acutely ill patients in Kenya was enriched and sequenced. Six new dengue-2 genomes were combined with 349 publicly available genomes and phylogenies used to infer gene flow between Kenya and other countries. Analyses indicate two dengue-2 Cosmopolitan genotype lineages circulating in Kenya, linked to recent outbreaks in coastal Kenya and Burkina Faso. Lineages circulating in Western, Southern, and Eastern Africa exhibiting similar evolutionary features are also reported. Phylogeography suggests importation of dengue-2 into Kenya from East and Southeast Asia and bidirectional geneflow. Additional lineages circulating in Africa are also imported from East and Southeast Asia. These findings underscore how intermittent importations from East and Southeast Asia drive dengue-2 circulation in Kenya and Africa more broadly.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
