

DrugSynthMC: An Atom-Based Generation of Drug-like Molecules with Monte Carlo Search
- PMID: 39249497
- PMCID: PMC11423341
- DOI: 10.1021/acs.jcim.4c01451
DrugSynthMC: An Atom-Based Generation of Drug-like Molecules with Monte Carlo Search
Abstract
A growing number of deep learning (DL) methodologies have recently been developed to design novel compounds and expand the chemical space within virtual libraries. Most of these neural network approaches design molecules to specifically bind a target based on its structural information and/or knowledge of previously identified binders. Fewer attempts have been made to develop approaches for de novo design of virtual libraries, as synthesizability of generated molecules remains a challenge. In this work, we developed a new Monte Carlo Search (MCS) algorithm, DrugSynthMC (Dru
Conflict of interest statement
The authors declare no competing financial interest.
Figures
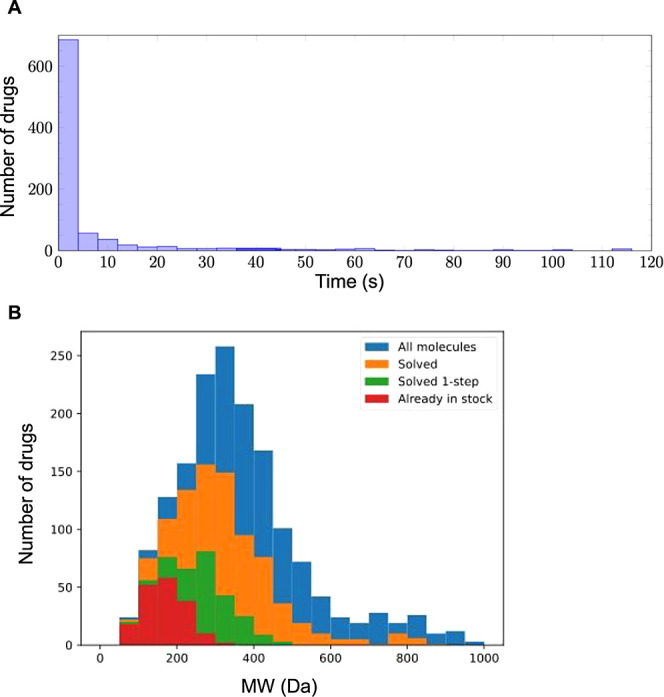
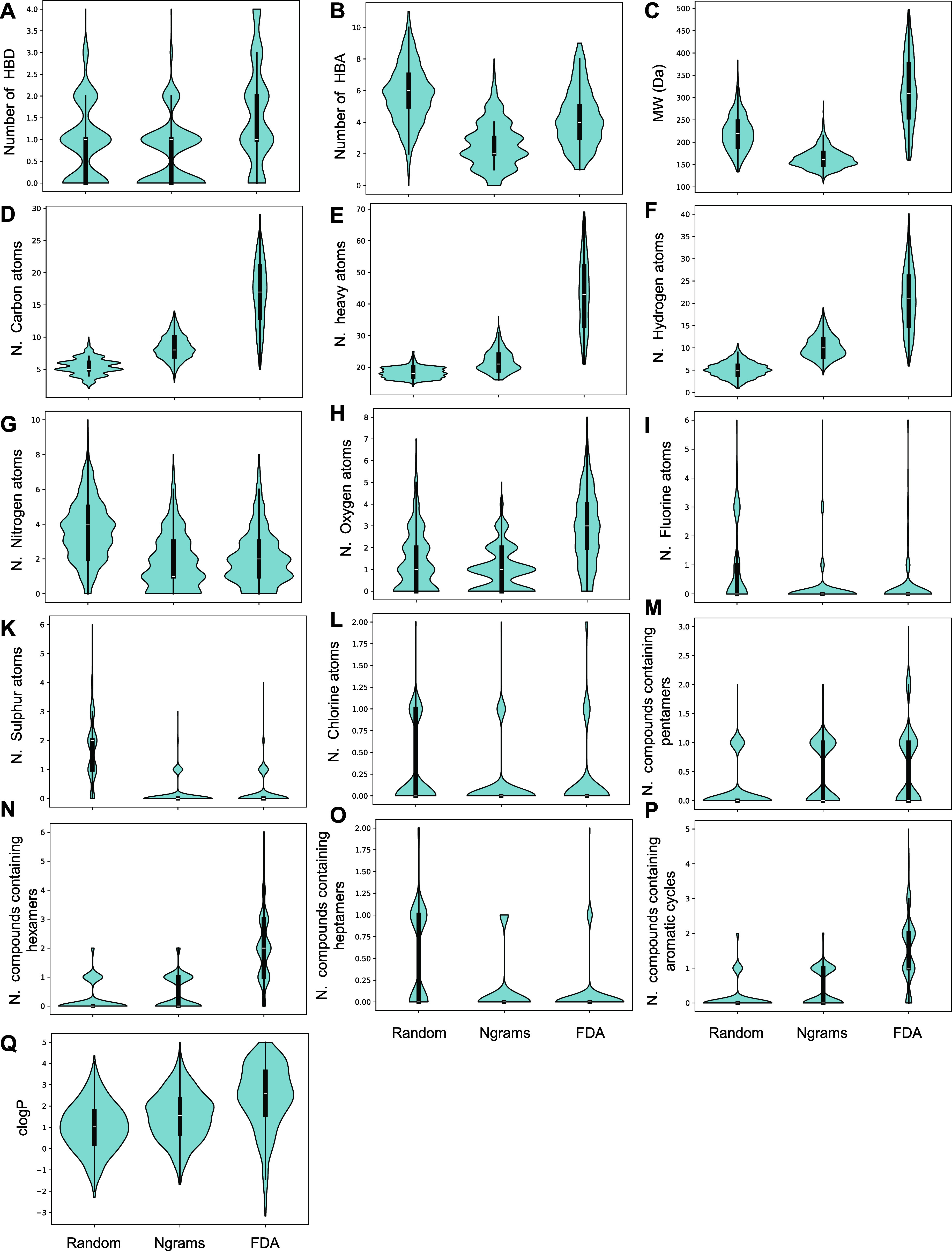
Similar articles
-
Target-specific novel molecules with their recipe: Incorporating synthesizability in the design process.J Mol Graph Model. 2024 Jun;129:108734. doi: 10.1016/j.jmgm.2024.108734. Epub 2024 Feb 28. J Mol Graph Model. 2024. PMID: 38442440
-
FSM-DDTR: End-to-end feedback strategy for multi-objective De Novo drug design using transformers.Comput Biol Med. 2023 Sep;164:107285. doi: 10.1016/j.compbiomed.2023.107285. Epub 2023 Jul 31. Comput Biol Med. 2023. PMID: 37557054
-
Toward efficient generation, correction, and properties control of unique drug-like structures.J Comput Chem. 2021 Apr 30;42(11):746-760. doi: 10.1002/jcc.26494. Epub 2021 Feb 14. J Comput Chem. 2021. PMID: 33583075
-
RetroGNN: Fast Estimation of Synthesizability for Virtual Screening and De Novo Design by Learning from Slow Retrosynthesis Software.J Chem Inf Model. 2022 May 23;62(10):2293-2300. doi: 10.1021/acs.jcim.1c01476. Epub 2022 Apr 22. J Chem Inf Model. 2022. PMID: 35452226 Review.
-
Exploration of Ultralarge Compound Collections for Drug Discovery.J Chem Inf Model. 2022 May 9;62(9):2021-2034. doi: 10.1021/acs.jcim.2c00224. Epub 2022 Apr 14. J Chem Inf Model. 2022. PMID: 35421301 Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
