

Glioblastoma cells increase expression of notch signaling and synaptic genes within infiltrated brain tissue
- PMID: 39251578
- PMCID: PMC11385527
- DOI: 10.1038/s41467-024-52167-y
Glioblastoma cells increase expression of notch signaling and synaptic genes within infiltrated brain tissue
Abstract
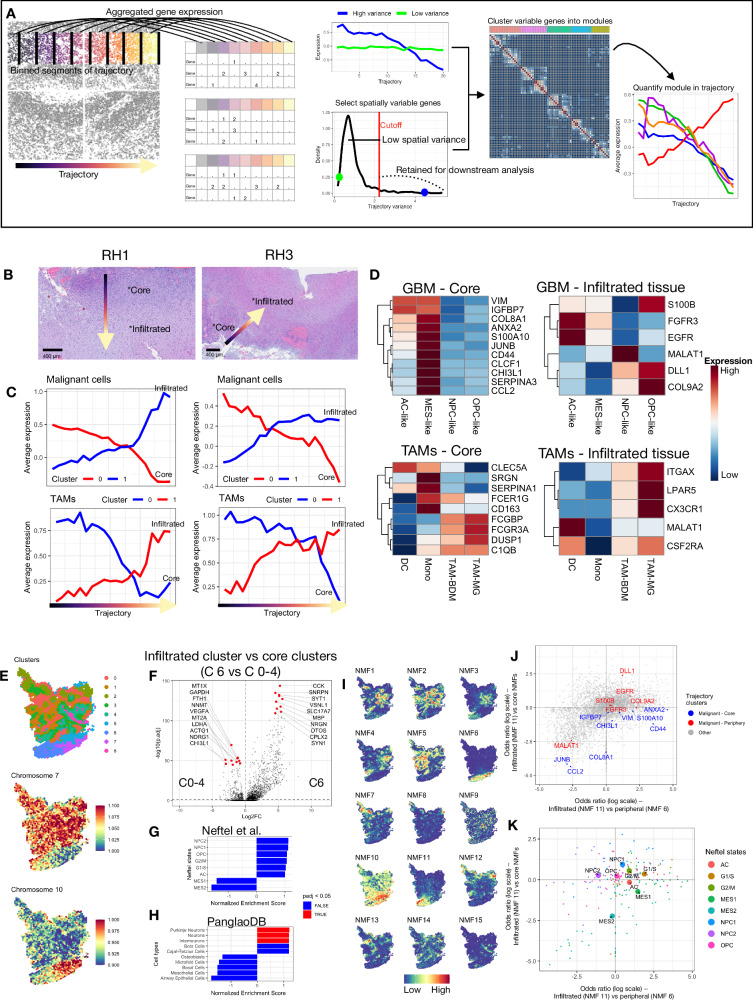
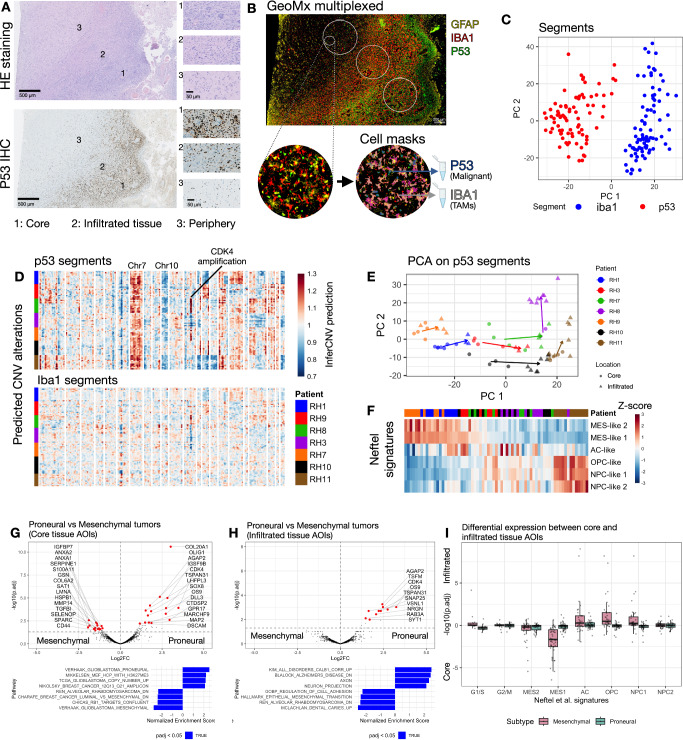
Glioblastoma remains one of the deadliest brain malignancies. First-line therapy consists of maximal surgical tumor resection, accompanied by chemotherapy and radiotherapy. Malignant cells escape surgical resection by migrating into the surrounding healthy brain tissue, where they give rise to the recurrent tumor. Based on gene expression, tumor cores can be subtyped into mesenchymal, proneural, and classical tumors, each being associated with differences in genetic alterations and cellular composition. In contrast, the adjacent brain parenchyma where infiltrating malignant cells escape surgical resection is less characterized in patients. Using spatial transcriptomics (n = 11), we show that malignant cells within proneural or mesenchymal tumor cores display spatially organized differences in gene expression, although such differences decrease within the infiltrated brain tissue. Malignant cells residing in infiltrated brain tissue have increased expression of genes related to neurodevelopmental pathways and glial cell differentiation. Our findings provide an updated view of the spatial landscape of glioblastomas and further our understanding of the malignant cells that infiltrate the healthy brain, providing new avenues for the targeted therapy of these cells after surgical resection.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
