

This is a preprint.
Temporally discordant chromatin accessibility and DNA demethylation define short and long-term enhancer regulation during cell fate specification
- PMID: 39253426
- PMCID: PMC11383056
- DOI: 10.1101/2024.08.27.609789
Temporally discordant chromatin accessibility and DNA demethylation define short and long-term enhancer regulation during cell fate specification
Update in
-
Temporally discordant chromatin accessibility and DNA demethylation define short- and long-term enhancer regulation during cell fate specification.Cell Rep. 2025 May 27;44(5):115680. doi: 10.1016/j.celrep.2025.115680. Epub 2025 May 9. Cell Rep. 2025. PMID: 40349339 Free PMC article.
Abstract
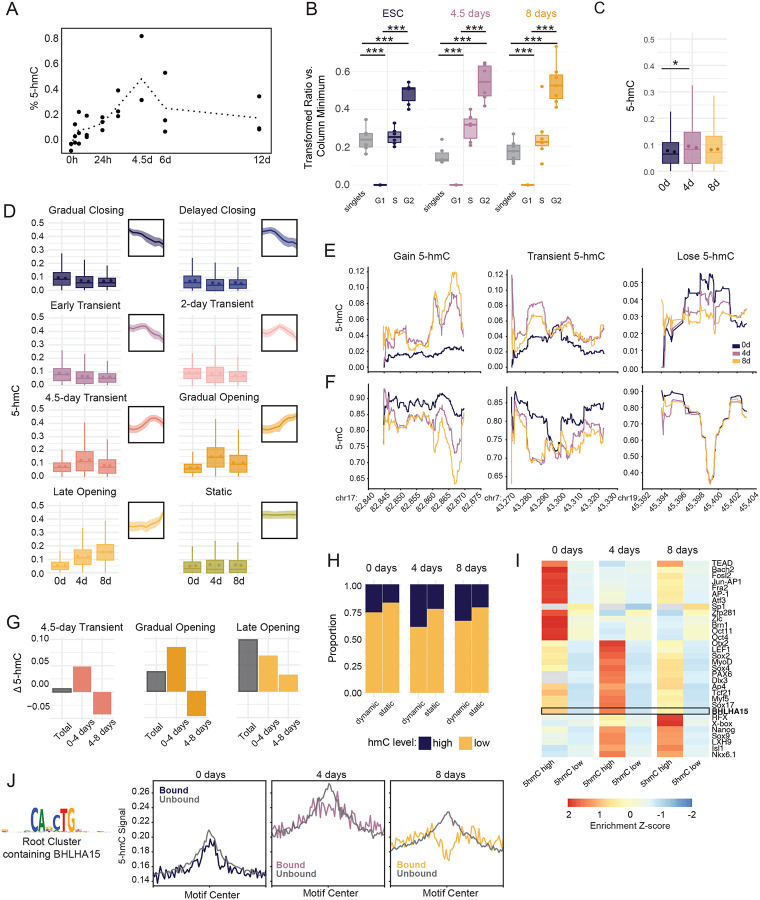
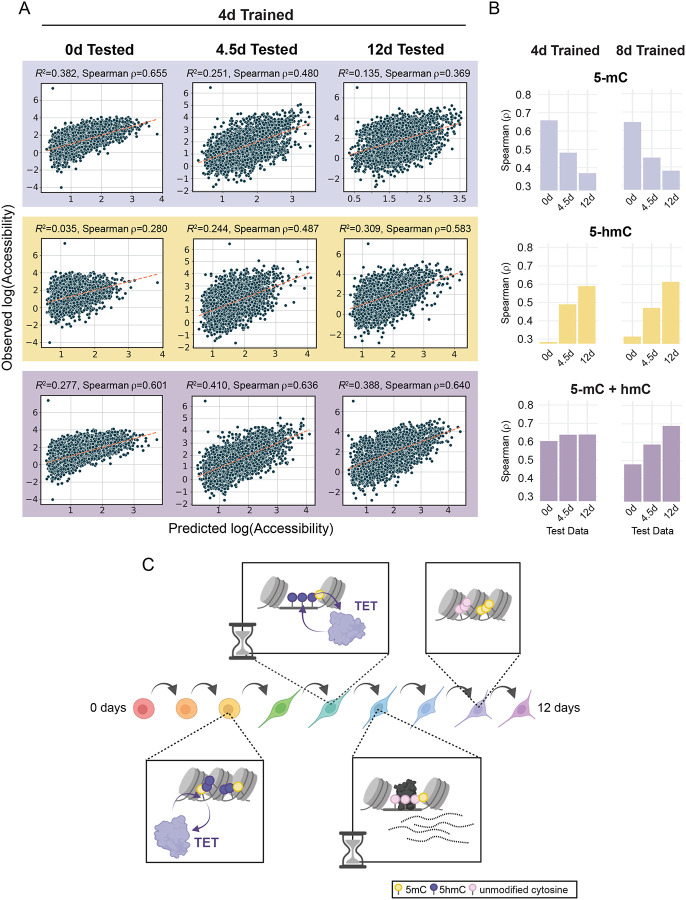
Epigenetic mechanisms govern the transcriptional activity of lineage-specifying enhancers; but recent work challenges the dogma that joint chromatin accessibility and DNA demethylation are prerequisites for transcription. To understand this paradox, we established a highly-resolved timeline of DNA demethylation, chromatin accessibility, and transcription factor occupancy during neural progenitor cell differentiation. We show thousands of enhancers undergo rapid, transient accessibility changes associated with distinct periods of transcription factor expression. However, most DNA methylation changes are unidirectional and delayed relative to chromatin dynamics, creating transiently discordant epigenetic states. Genome-wide detection of 5-hydroxymethylcytosine further revealed active demethylation begins ahead of chromatin and transcription factor activity, while enhancer hypomethylation persists long after these activities have dissipated. We demonstrate that these timepoint specific methylation states predict past, present and future chromatin accessibility using machine learning models. Thus, chromatin and DNA methylation collaborate on different timescales to mediate short and long-term enhancer regulation during cell fate specification.
Keywords: 5-hydroxymethylation; 6-base sequencing; ATAC-Me; Chromatin Accessibility; DNA Methylation; Differentiation; Enhancers; Epigenetics; Machine Learning; Neural Progenitor Cells.
Conflict of interest statement
DECLARATION OF INTERESTS F.P., A.J., and T.C. are employees of biomodal, formerly Cambridge Epigenetix. All other authors declare no competing interests.
Figures
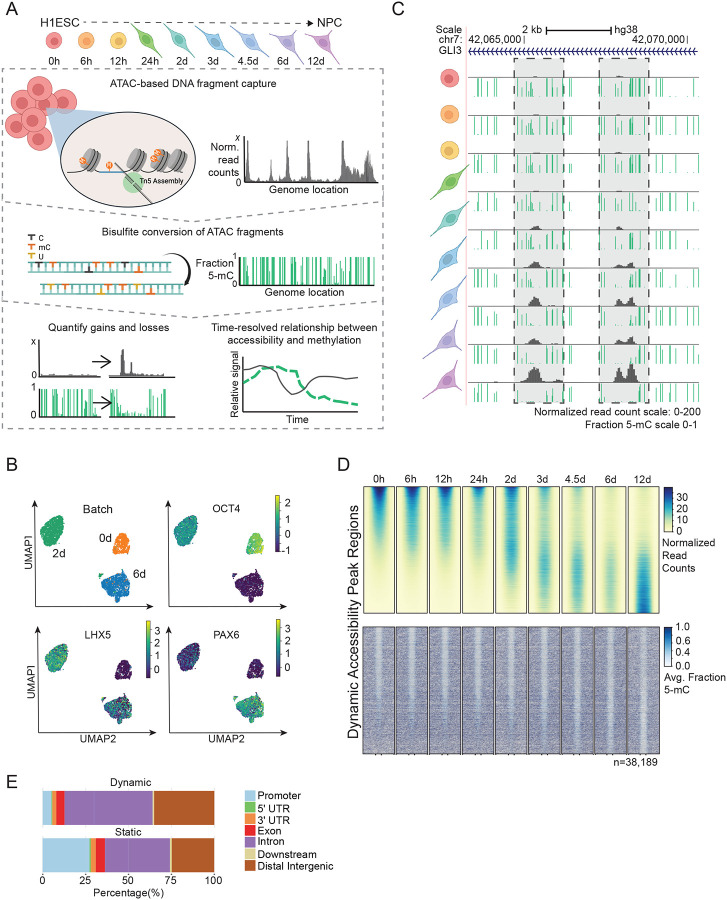
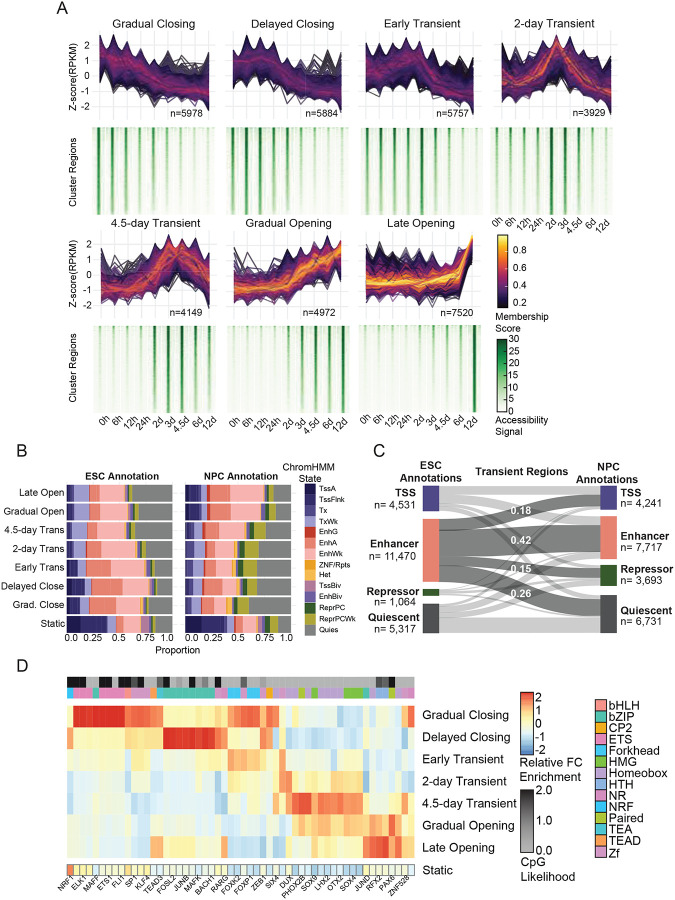
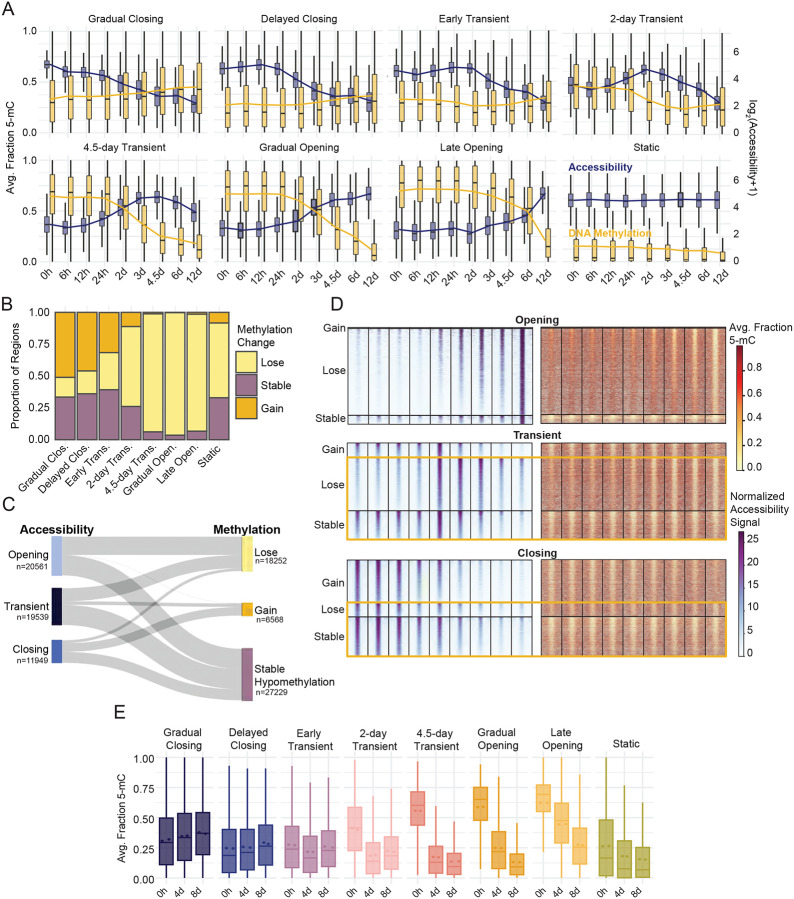
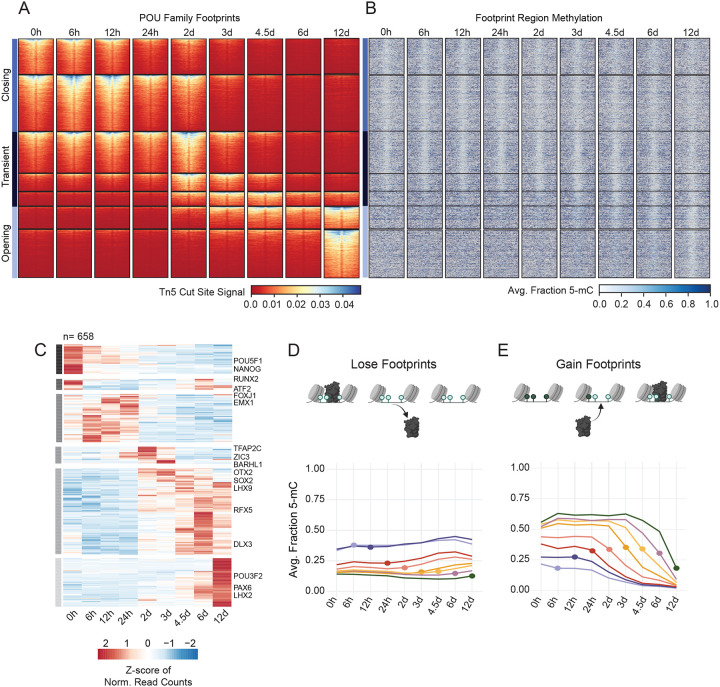
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous