

This is a preprint.
Common DNA sequence variation influences epigenetic aging in African populations
- PMID: 39253488
- PMCID: PMC11383046
- DOI: 10.1101/2024.08.26.608843
Common DNA sequence variation influences epigenetic aging in African populations
Update in
-
Common DNA sequence variation influences epigenetic aging in African populations.Commun Biol. 2025 Nov 5;8(1):1530. doi: 10.1038/s42003-025-08893-0. Commun Biol. 2025. PMID: 41193633 Free PMC article.
Abstract
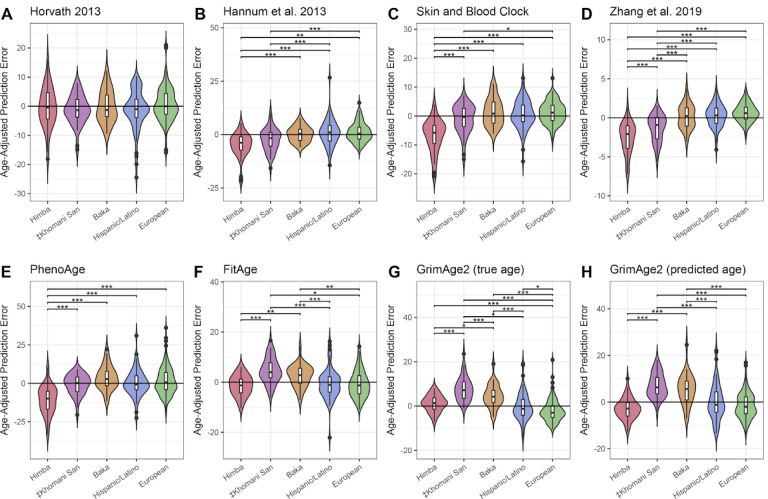
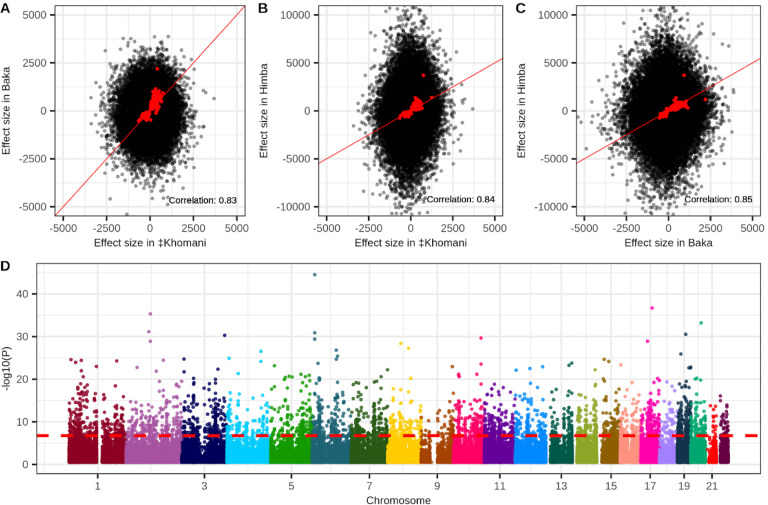
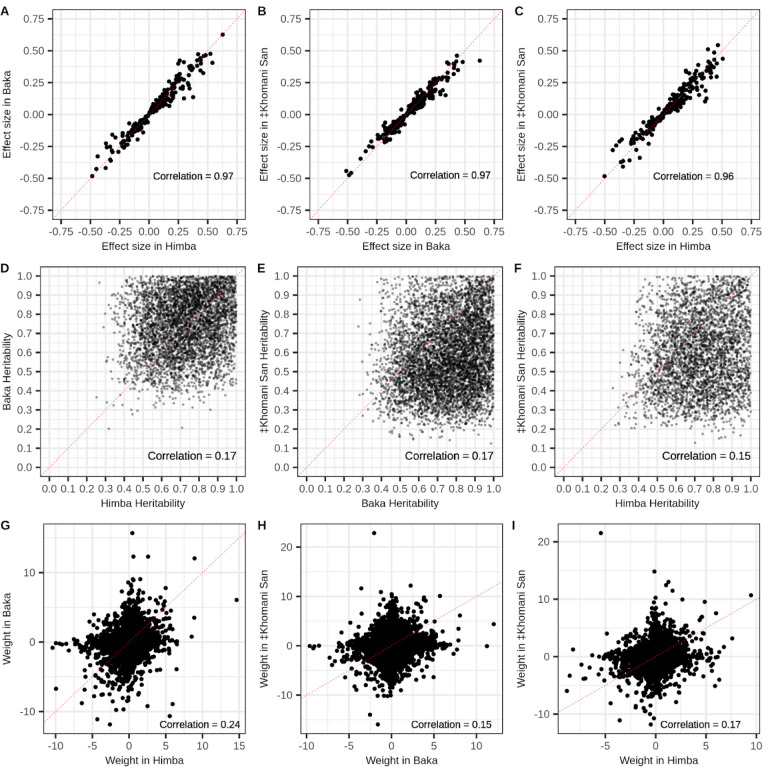
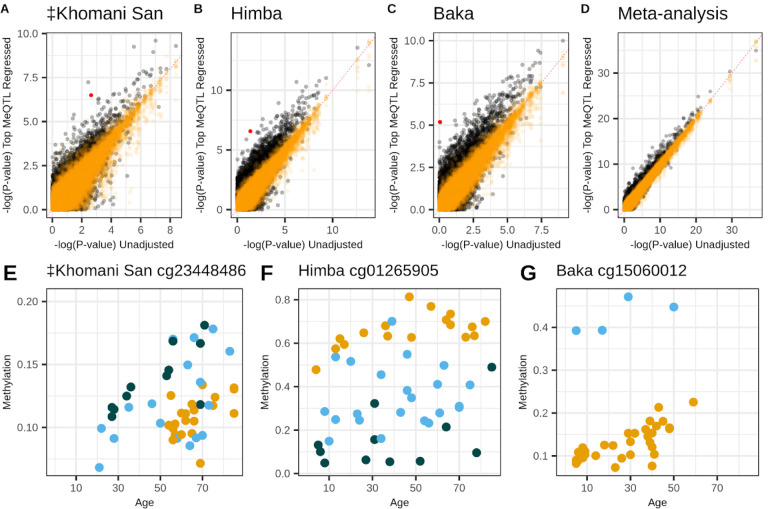
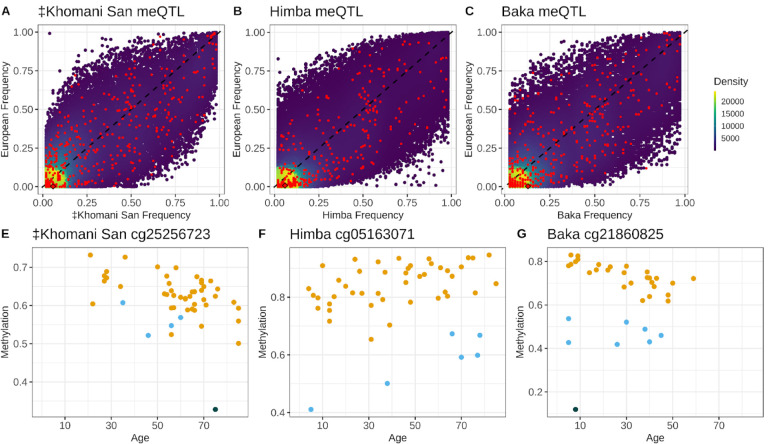
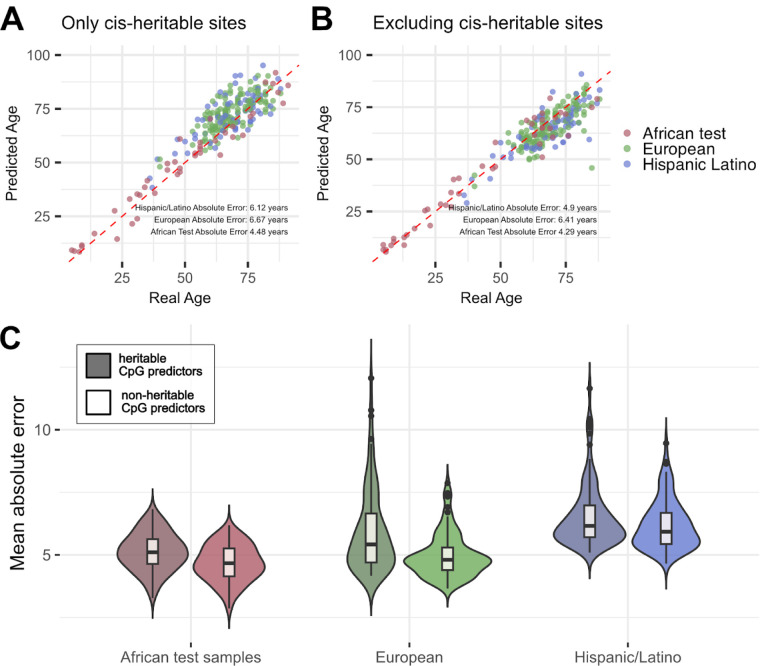
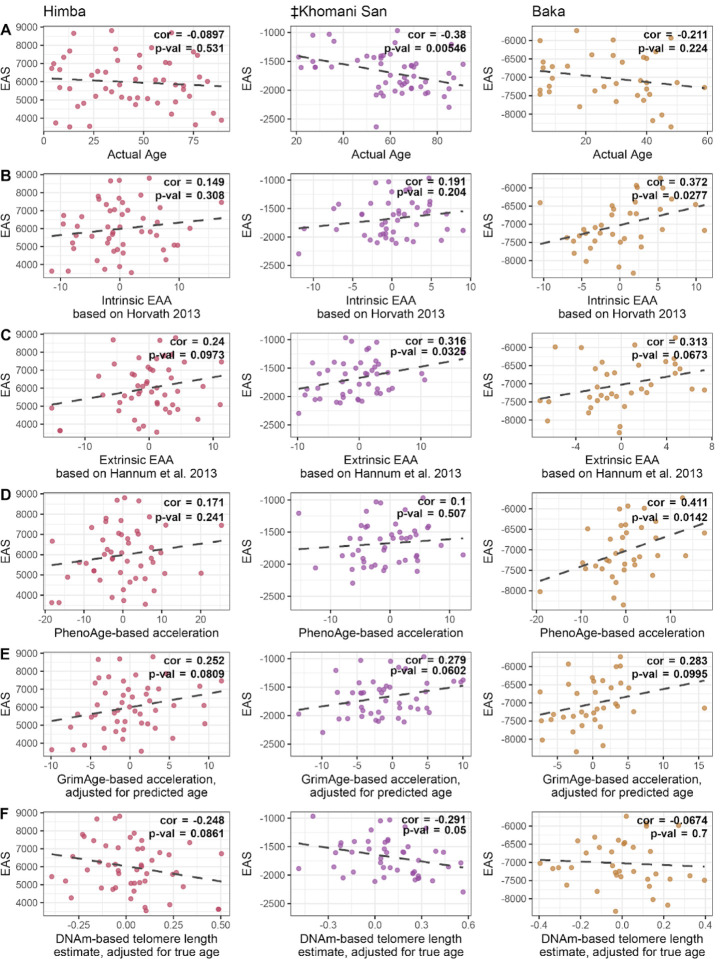
Aging is associated with genome-wide changes in DNA methylation in humans, facilitating the development of epigenetic age prediction models. However, most of these models have been trained primarily on European-ancestry individuals, and none account for the impact of methylation quantitative trait loci (meQTL). To address these gaps, we analyzed the relationships between age, genotype, and CpG methylation in 3 understudied populations: central African Baka (n = 35), southern African ‡Khomani San (n = 52), and southern African Himba (n = 51). We find that published prediction methods yield higher mean errors in these cohorts compared to European-ancestry individuals, and find that unaccounted-for DNA sequence variation may be a significant factor underlying this loss of accuracy. We leverage information about the associations between DNA genotype and CpG methylation to develop an age predictor that is minimally influenced by meQTL, and show that this model remains accurate across a broad range of genetic backgrounds. Intriguingly, we also find that the older individuals and those exhibiting relatively lower epigenetic age acceleration in our cohorts tend to carry more epigenetic age-reducing genetic variants, suggesting a novel mechanism by which heritable factors can influence longevity.
Conflict of interest statement
Competing Interests The authors have no competing interests to declare.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases