

Lethal Borna disease virus 1 infections of humans and animals - in-depth molecular epidemiology and phylogeography
- PMID: 39256401
- PMCID: PMC11387626
- DOI: 10.1038/s41467-024-52192-x
Lethal Borna disease virus 1 infections of humans and animals - in-depth molecular epidemiology and phylogeography
Abstract
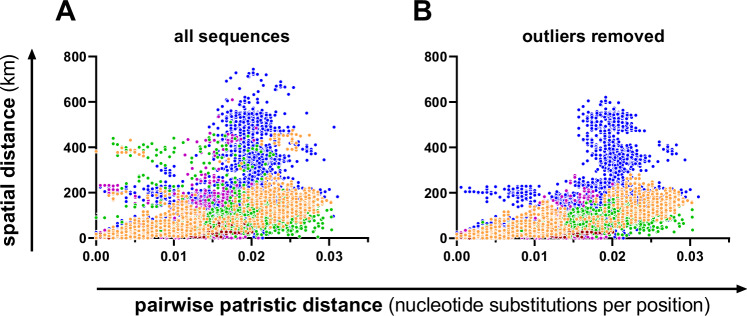
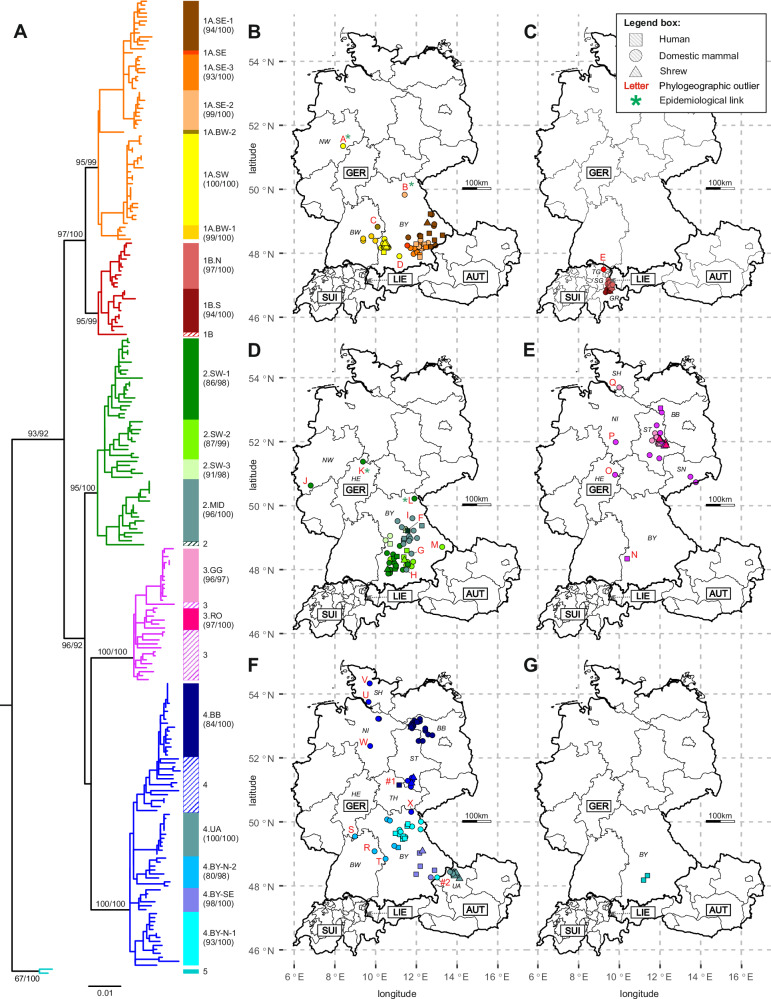
Borna disease virus 1 (BoDV-1) is the causative agent of Borna disease, a fatal neurologic disorder of domestic mammals and humans, resulting from spill-over infection from its natural reservoir host, the bicolored white-toothed shrew (Crocidura leucodon). The known BoDV-1-endemic area is remarkably restricted to parts of Germany, Austria, Switzerland and Liechtenstein. To gain comprehensive data on its occurrence, we analysed diagnostic material from suspected BoDV-1-induced encephalitis cases based on clinical and/or histopathological diagnosis. BoDV-1 infection was confirmed by RT-qPCR in 207 domestic mammals, 28 humans and seven wild shrews. Thereby, this study markedly raises the number of published laboratory-confirmed human BoDV-1 infections and provides a first comprehensive summary. Generation of 136 new BoDV-1 genome sequences from animals and humans facilitated an in-depth phylogeographic analysis, allowing for the definition of risk areas for zoonotic BoDV-1 transmission and facilitating the assessment of geographical infection sources. Consistent with the low mobility of its reservoir host, BoDV-1 sequences showed a remarkable geographic association, with individual phylogenetic clades occupying distinct areas. The closest genetic relatives of most human-derived BoDV-1 sequences were located at distances of less than 40 km, indicating that spill-over transmission from the natural reservoir usually occurs in the patient´s home region.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Dürrwald, R., Nowotny, N., Beer, M. & Kuhn JH. Clinical Virology (eds Richman D. D., Whitley R. J., Hayden F. G.). 4th edn. (American Society for Microbiology, 2016).
Publication types
MeSH terms
Grants and funding
- 01KI1722/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- 01KI2005/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- 01KI1903B/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- 01KI2002/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- 504757758/Deutsche Forschungsgemeinschaft (German Research Foundation)
LinkOut - more resources
Full Text Sources
