

Targeting Rap1b signaling cascades with CDNF: Mitigating platelet activation, plasma oxylipins and reperfusion injury in stroke
- PMID: 39256999
- PMCID: PMC11573613
- DOI: 10.1016/j.ymthe.2024.09.005
Targeting Rap1b signaling cascades with CDNF: Mitigating platelet activation, plasma oxylipins and reperfusion injury in stroke
Abstract
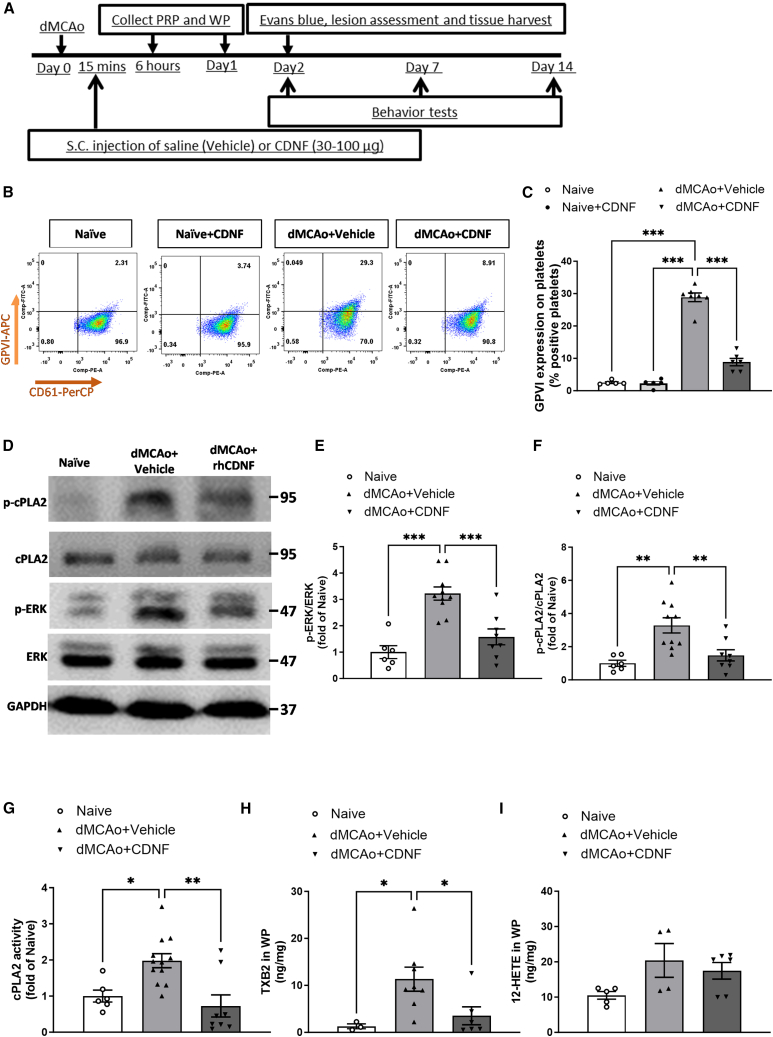
Cerebral reperfusion injury in stroke, stemming from interconnected thrombotic and inflammatory signatures, often involves platelet activation, aggregation and its interaction with various immune cells, contributing to microvascular dysfunction. However, the regulatory mechanisms behind this platelet activation and the resulting inflammation are not well understood, complicating the development of effective stroke therapies. Utilizing animal models and platelets from hemorrhagic stroke patients, our research demonstrates that human cerebral dopamine neurotrophic factor (CDNF) acts as an endogenous antagonist, mitigating platelet aggregation and associated neuroinflammation. CDNF moderates mitochondrial membrane potential, reactive oxygen species production, and intracellular calcium in activated platelets by interfering with GTP binding to Rap1b, thereby reducing Rap1b activation and downregulating the Rap1b-MAPK-PLA2 signaling pathway, which decreases release of the pro-inflammatory mediator thromboxane A2. In addition, CDNF reduces the inflammatory response in BV2 microglial cells co-cultured with activated platelets. Consistent with ex vivo findings, subcutaneous administration of CDNF in a rat model of ischemic stroke significantly reduces platelet activation, aggregation, lipid mediator production, infarct volume, and neurological deficits. In summary, our study highlights CDNF as a promising therapeutic target for mitigating platelet-induced inflammation and enhancing recovery in stroke. Harnessing the CDNF pathway may offer a novel therapeutic strategy for stroke intervention.
Keywords: CDNF; Rap1b; cerebral dopamine neurotrophic factor; dMCAo; distal middle cerebral artery occlusion; ischemic stroke; oxylipin metabolism; platelet activation.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.S. is one of inventors of the CDNF-related patent (7452969), which is owned by the Herantis Pharma Company (Espoo, Finland).
Figures








References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
