

Familial severe skeletal Class II malocclusion with gingival hyperplasia caused by a complex structural rearrangement at the KCNJ2-KCNJ16 locus
- PMID: 39257002
- PMCID: PMC11465088
- DOI: 10.1016/j.xhgg.2024.100352
Familial severe skeletal Class II malocclusion with gingival hyperplasia caused by a complex structural rearrangement at the KCNJ2-KCNJ16 locus
Abstract
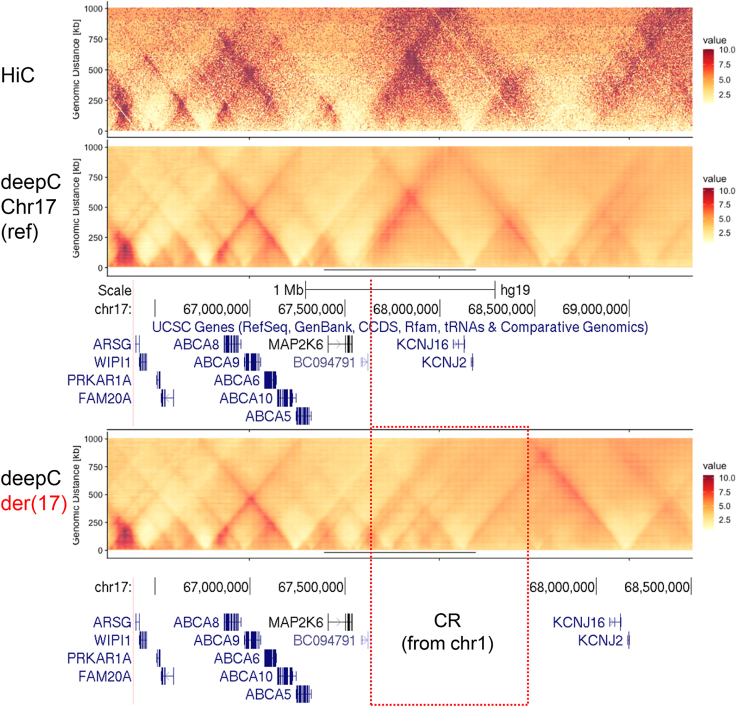
The aim of this work was to identify the underlying genetic cause in a four-generation family segregating an unusual phenotype comprising a severe form of skeletal Class II malocclusion with gingival hyperplasia. SNP array identified a copy number gain on chromosome 1 (chr1); however, this chromosomal region did not segregate correctly in the extended family. Exome sequencing also failed to identify a candidate causative variant but highlighted co-segregating genetic markers on chr17 and chr19. Short- and long-read genome sequencing allowed us to pinpoint and characterize at nucleotide-level resolution a chromothripsis-like complex rearrangement (CR) inserted into the chr17 co-segregating region at the KCNJ2-SOX9 locus. The CR involved the gain of five different regions from chr1 that are shuffled, chained, and inserted as a single block (∼828 kb) at chr17q24.3. The inserted sequences contain craniofacial enhancers that are predicted to interact with KCNJ2/KCNJ16 through neo-topologically associating domain (TAD) formation to induce ectopic activation. Our findings suggest that the CR inserted at chr17q24.3 is the cause of the severe skeletal Class II malocclusion with gingival hyperplasia in this family and expands the panoply of phenotypes linked to variation at the KCNJ2-SOX9 locus. In addition, we highlight a previously overlooked potential role for misregulation of the KCNJ2/KCNJ16 genes in the pathomechanism of gingival hyperplasia associated with deletions and other rearrangements of the 17q24.2-q24.3 region (MIM 135400).
Keywords: KCNJ16; KCNJ2; chromoanagenesis; chromothripsis; enhancer hijacking; gingival hyperplasia; malocclusion; mandibular retrognathism; maxillary prognathism; maxillary protrusion; neo-TAD.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
