

This is a preprint.
Computational design of serine hydrolases
- PMID: 39257749
- PMCID: PMC11384011
- DOI: 10.1101/2024.08.29.610411
Computational design of serine hydrolases
Update in
-
Computational design of serine hydrolases.Science. 2025 Apr 18;388(6744):eadu2454. doi: 10.1126/science.adu2454. Epub 2025 Apr 18. Science. 2025. PMID: 39946508 Free PMC article.
Abstract
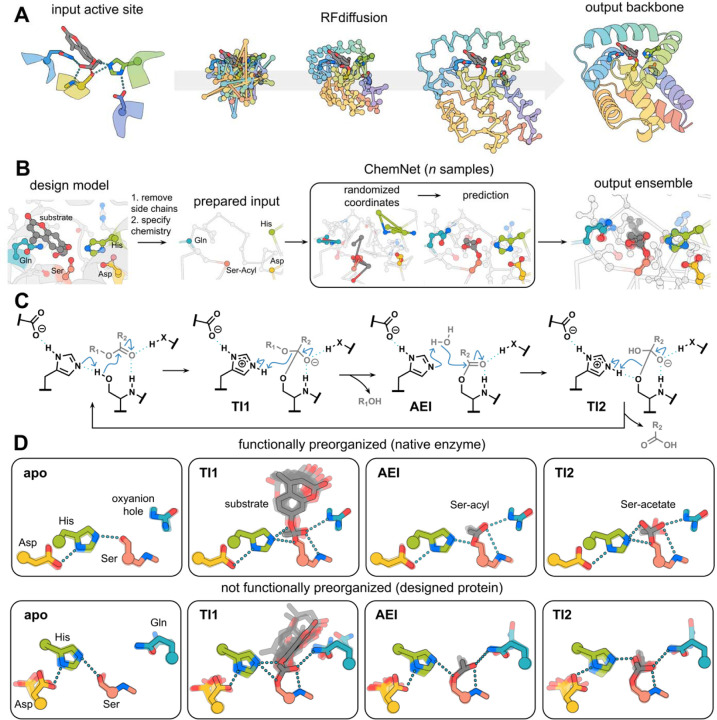
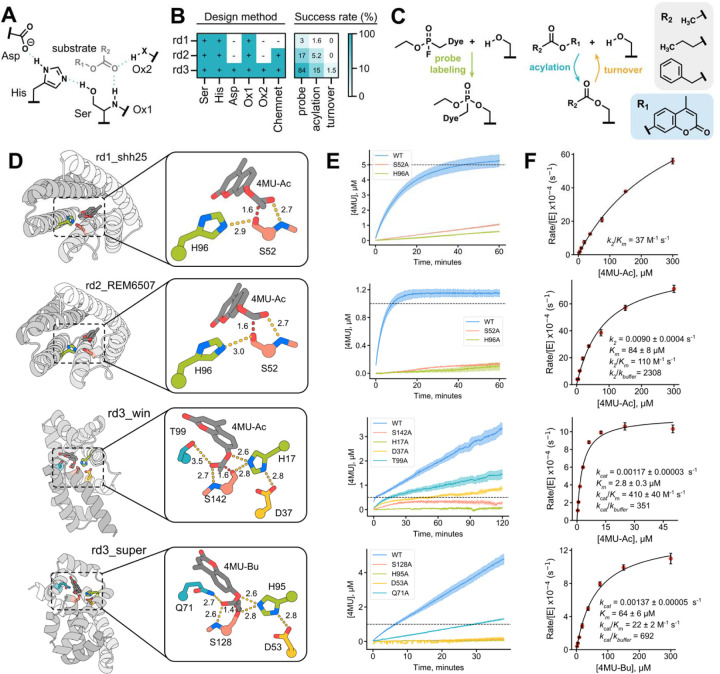
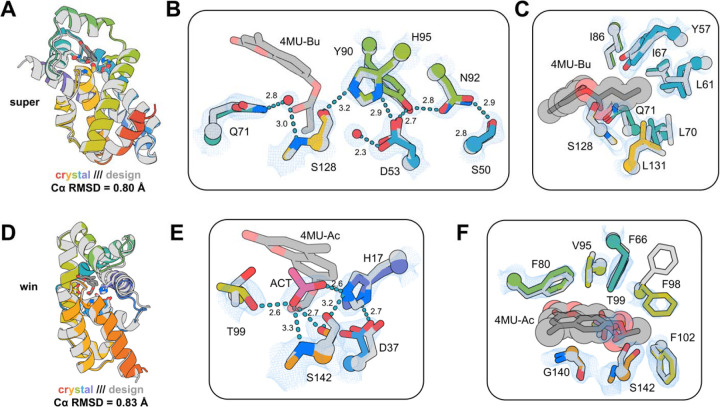
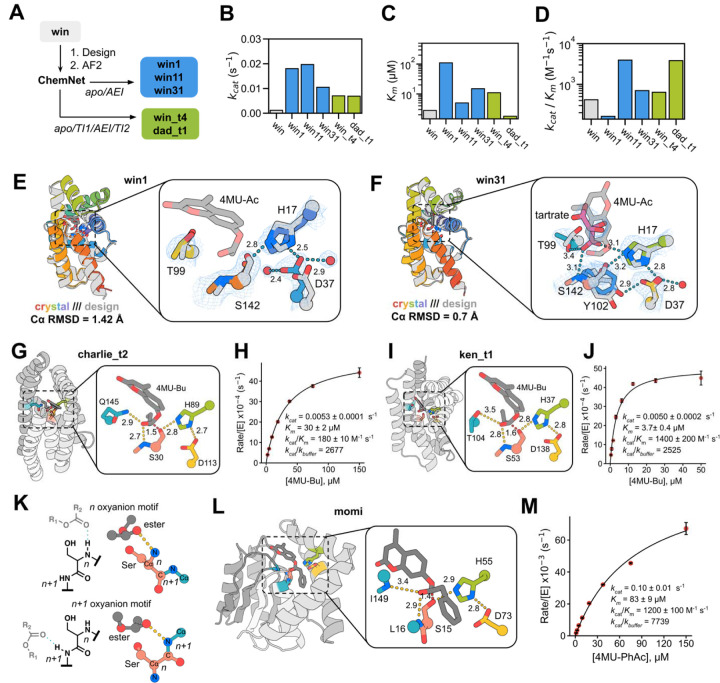
Enzymes that proceed through multistep reaction mechanisms often utilize complex, polar active sites positioned with sub-angstrom precision to mediate distinct chemical steps, which makes their de novo construction extremely challenging. We sought to overcome this challenge using the classic catalytic triad and oxyanion hole of serine hydrolases as a model system. We used RFdiffusion1 to generate proteins housing catalytic sites of increasing complexity and varying geometry, and a newly developed ensemble generation method called ChemNet to assess active site geometry and preorganization at each step of the reaction. Experimental characterization revealed novel serine hydrolases that catalyze ester hydrolysis with catalytic efficiencies (k cat /K m ) up to 3.8 x 103 M-1 s-1, closely match the design models (Cα RMSDs < 1 Å), and have folds distinct from natural serine hydrolases. In silico selection of designs based on active site preorganization across the reaction coordinate considerably increased success rates, enabling identification of new catalysts in screens of as few as 20 designs. Our de novo buildup approach provides insight into the geometric determinants of catalysis that complements what can be obtained from structural and mutational studies of native enzymes (in which catalytic group geometry and active site makeup cannot be so systematically varied), and provides a roadmap for the design of industrially relevant serine hydrolases and, more generally, for designing complex enzymes that catalyze multi-step transformations.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous