

Enriching productive mutational paths accelerates enzyme evolution
- PMID: 39261644
- PMCID: PMC11581979
- DOI: 10.1038/s41589-024-01712-3
Enriching productive mutational paths accelerates enzyme evolution
Abstract
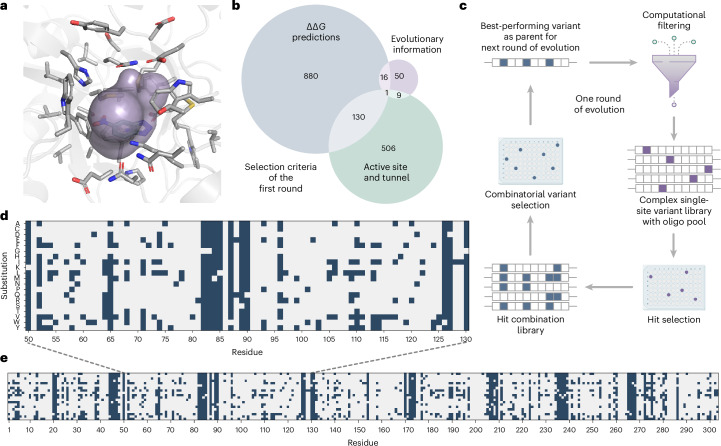
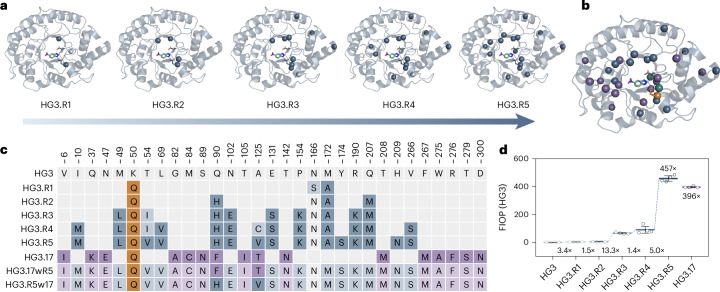
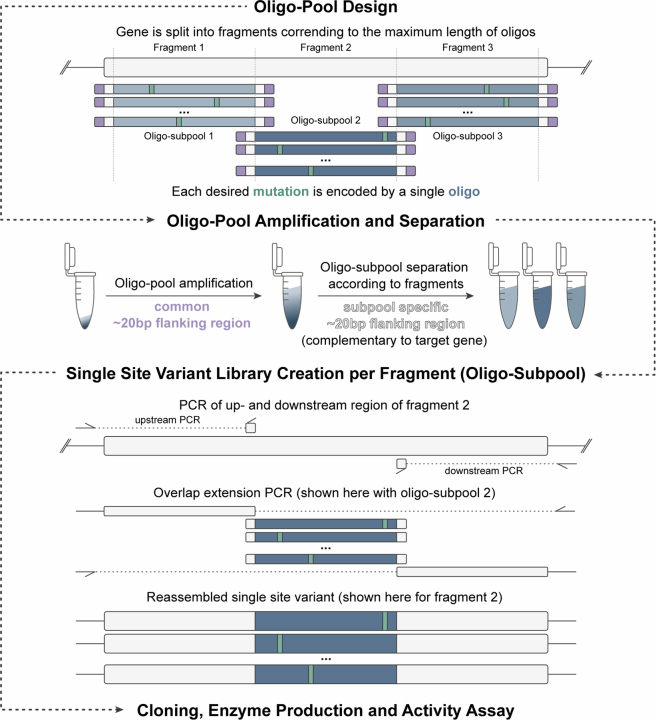
Darwinian evolution has given rise to all the enzymes that enable life on Earth. Mimicking natural selection, scientists have learned to tailor these biocatalysts through recursive cycles of mutation, selection and amplification, often relying on screening large protein libraries to productively modulate the complex interplay between protein structure, dynamics and function. Here we show that by removing destabilizing mutations at the library design stage and taking advantage of recent advances in gene synthesis, we can accelerate the evolution of a computationally designed enzyme. In only five rounds of evolution, we generated a Kemp eliminase-an enzymatic model system for proton transfer from carbon-that accelerates the proton abstraction step >108-fold over the uncatalyzed reaction. Recombining the resulting variant with a previously evolved Kemp eliminase HG3.17, which exhibits similar activity but differs by 29 substitutions, allowed us to chart the topography of the designer enzyme's fitness landscape, highlighting that a given protein scaffold can accommodate several, equally viable solutions to a specific catalytic problem.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: All authors declare no competing interests.
Figures








Similar articles
-
Precision is essential for efficient catalysis in an evolved Kemp eliminase.Nature. 2013 Nov 21;503(7476):418-21. doi: 10.1038/nature12623. Epub 2013 Oct 16. Nature. 2013. PMID: 24132235
-
Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series.J Mol Biol. 2010 Mar 5;396(4):1025-42. doi: 10.1016/j.jmb.2009.12.031. Epub 2009 Dec 28. J Mol Biol. 2010. PMID: 20036254
-
Natural Evolution Provides Strong Hints about Laboratory Evolution of Designer Enzymes.Proc Natl Acad Sci U S A. 2022 Aug 2;119(31):e2207904119. doi: 10.1073/pnas.2207904119. Epub 2022 Jul 28. Proc Natl Acad Sci U S A. 2022. PMID: 35901204 Free PMC article.
-
A roadmap to directed enzyme evolution and screening systems for biotechnological applications.Biol Res. 2013;46(4):395-405. doi: 10.4067/S0716-97602013000400011. Biol Res. 2013. PMID: 24510142 Review.
-
The role of reorganization energy in rational enzyme design.Curr Opin Chem Biol. 2014 Aug;21:34-41. doi: 10.1016/j.cbpa.2014.03.011. Epub 2014 Apr 24. Curr Opin Chem Biol. 2014. PMID: 24769299 Review.
Cited by
-
Multi-strategy ugt mining, modification and glycosyl donor synthesis facilitate the production of triterpenoid saponins.Front Plant Sci. 2025 May 30;16:1586295. doi: 10.3389/fpls.2025.1586295. eCollection 2025. Front Plant Sci. 2025. PMID: 40519597 Free PMC article. Review.
-
Emergence of specific binding and catalysis from a designed generalist binding protein.bioRxiv [Preprint]. 2025 Jul 22:2025.01.30.635804. doi: 10.1101/2025.01.30.635804. bioRxiv. 2025. PMID: 39975260 Free PMC article. Preprint.
-
Complete computational design of high-efficiency Kemp elimination enzymes.Nature. 2025 Jul;643(8074):1421-1427. doi: 10.1038/s41586-025-09136-2. Epub 2025 Jun 18. Nature. 2025. PMID: 40533551 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
