

IKKε-deficient macrophages impede cardiac repair after myocardial infarction by enhancing the macrophage-myofibroblast transition
- PMID: 39261656
- PMCID: PMC11446912
- DOI: 10.1038/s12276-024-01304-0
IKKε-deficient macrophages impede cardiac repair after myocardial infarction by enhancing the macrophage-myofibroblast transition
Abstract
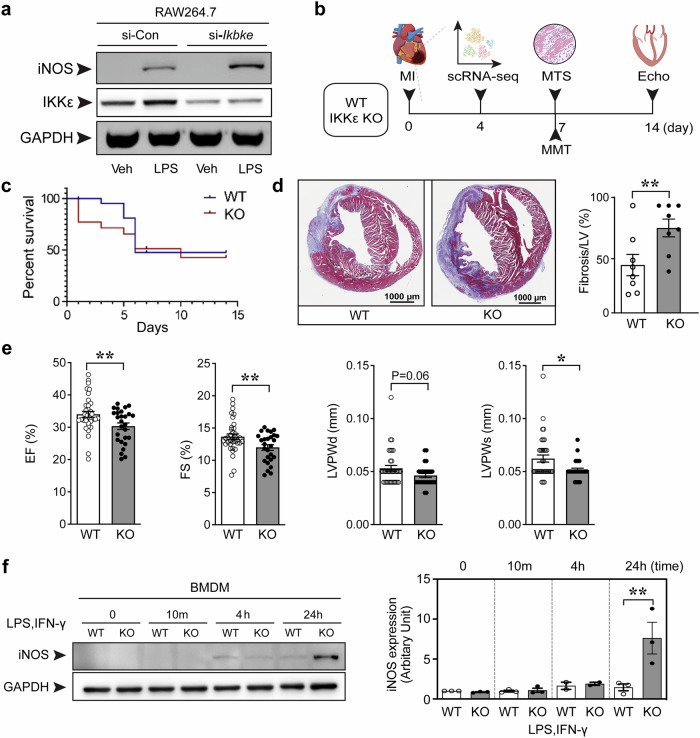
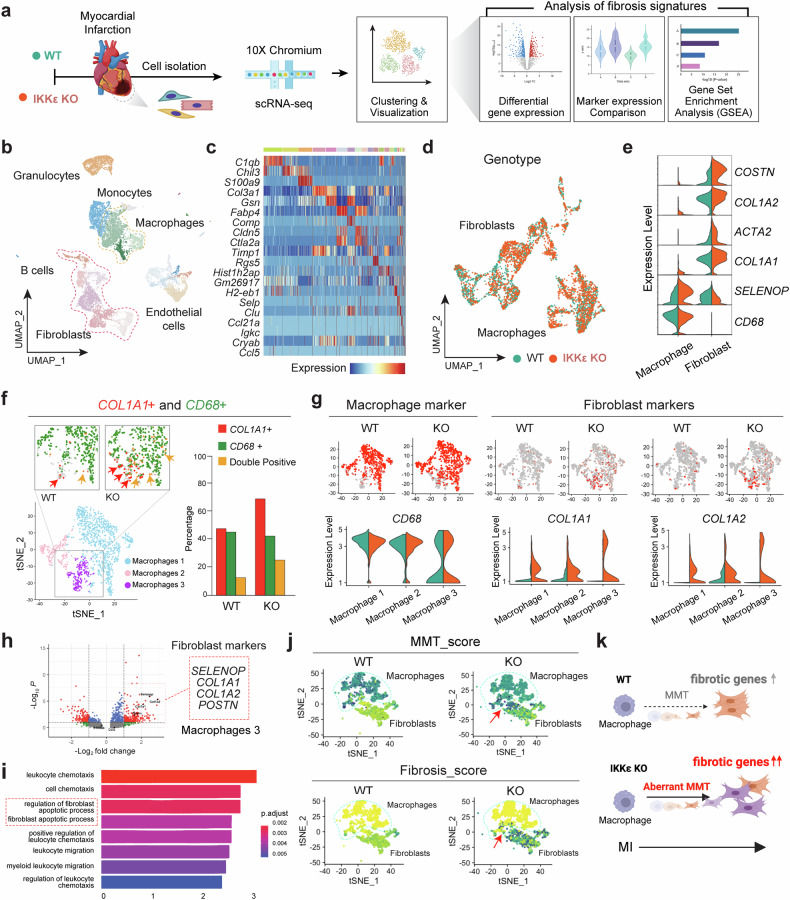
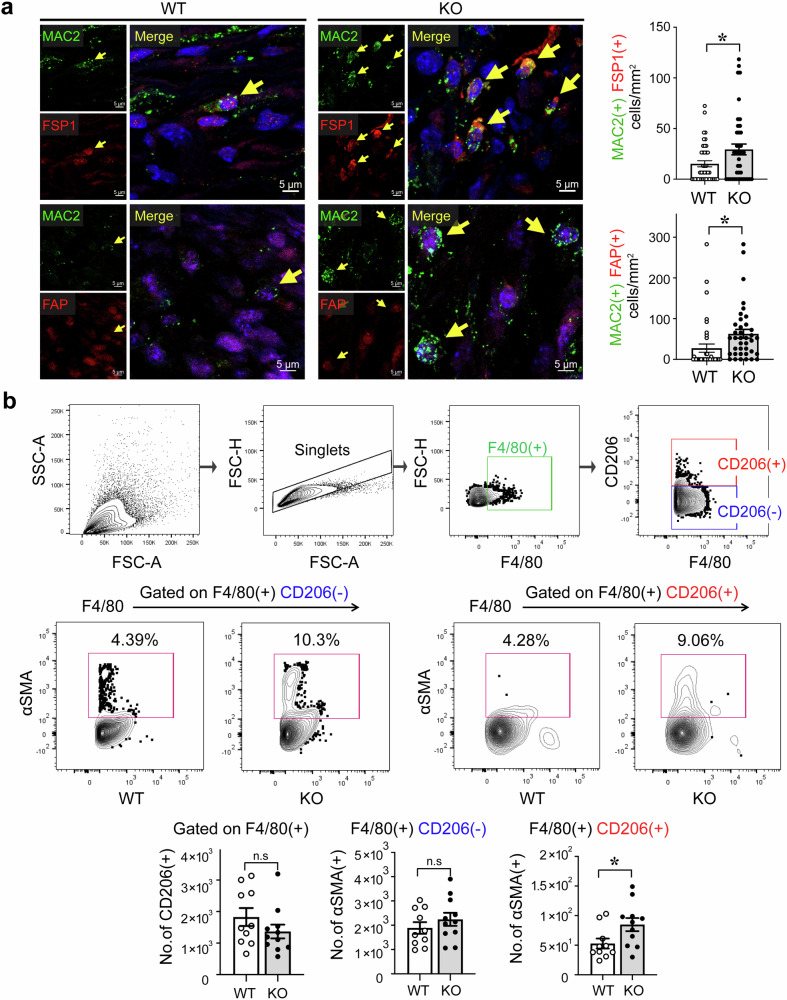
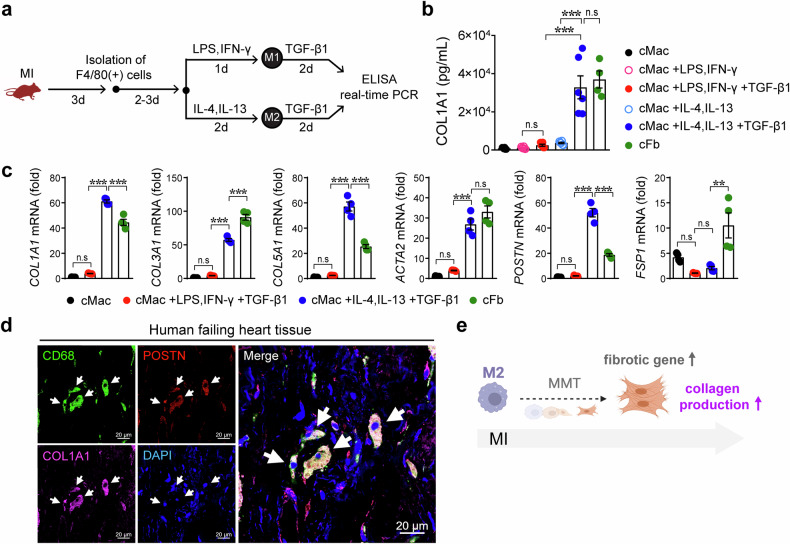
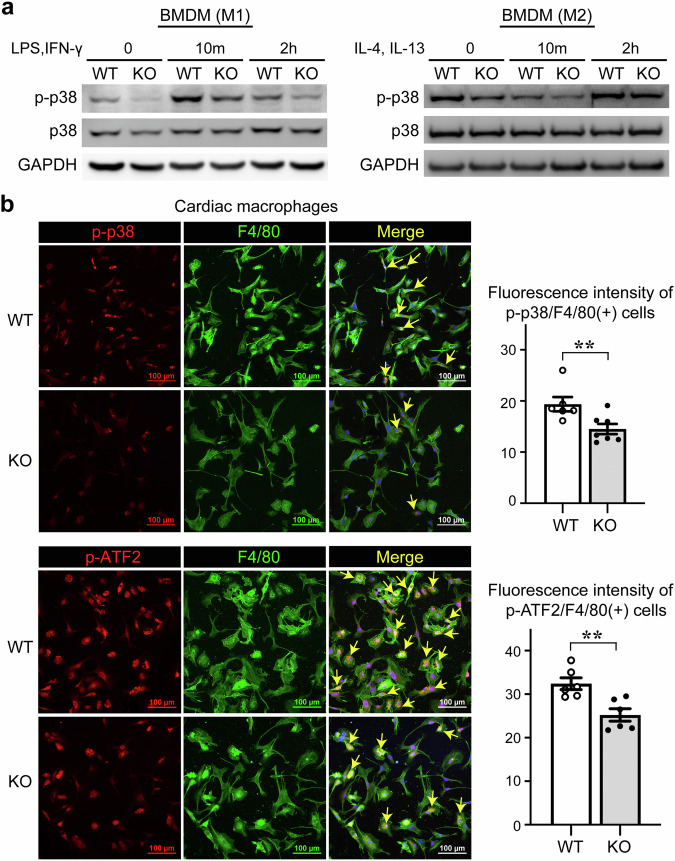
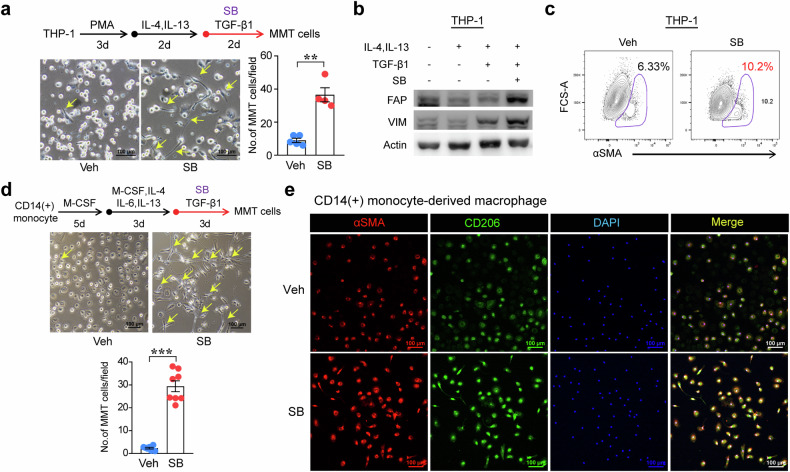
The regulatory role of the inhibitor of NF-kB kinase ε (IKKε) in postmyocardial infarction (MI) inflammation remains uncertain. Using an MI mouse model, we examined the cardiac outcomes of IKKε knockout (KO) mice and wild-type mice. We employed single-cell RNA sequencing (scRNA-seq) and phosphorylated protein array techniques to profile cardiac macrophages. IKKε KO mice exhibited compromised survival, heightened inflammation, pronounced cardiac fibrosis, and a reduced ejection fraction. A distinct cardiac macrophage subset in IKKε KO mice exhibited increased fibrotic marker expression and decreased phosphorylated p38 (p-p38) levels, indicating an enhanced macrophage-myofibroblast transition (MMT) post-MI. While cardiac inflammation is crucial for initiating compensatory pathways, the timely resolution of inflammation was impaired in the IKKε KO group, while the MMT in macrophages accelerated post-MI, leading to cardiac failure. Additionally, our study highlighted the potential of 5-azacytidine (5-Aza), known for its anti-inflammatory and cardioprotective effects, in restoring p-p38 levels in stimulated macrophages. The administration of 5-Aza significantly reduced the MMT in cardiac macrophages from the IKKε KO group. These findings underscore the regulation of the inflammatory response and macrophage transition by the IKKε-p38 axis, indicating that the MMT is a promising therapeutic target for ischemic heart disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
