

Disease prediction with multi-omics and biomarkers empowers case-control genetic discoveries in the UK Biobank
- PMID: 39261665
- PMCID: PMC11390475
- DOI: 10.1038/s41588-024-01898-1
Disease prediction with multi-omics and biomarkers empowers case-control genetic discoveries in the UK Biobank
Abstract
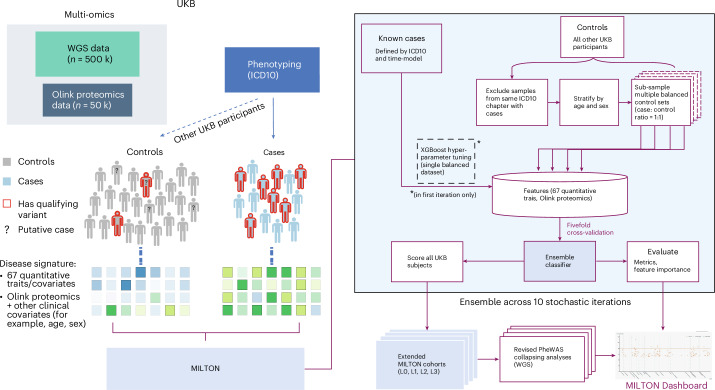
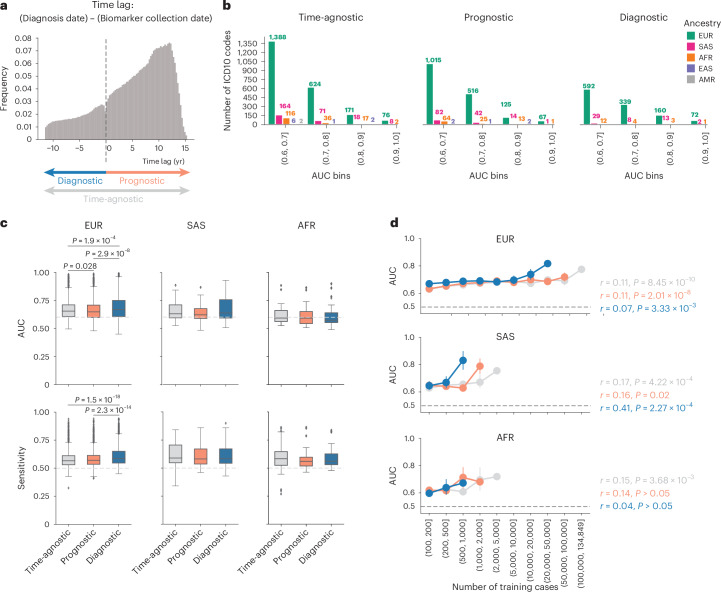
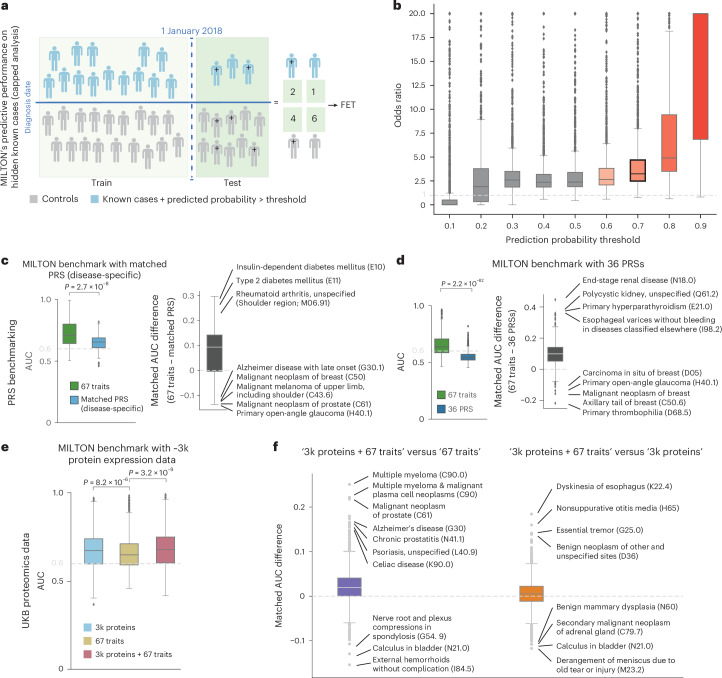
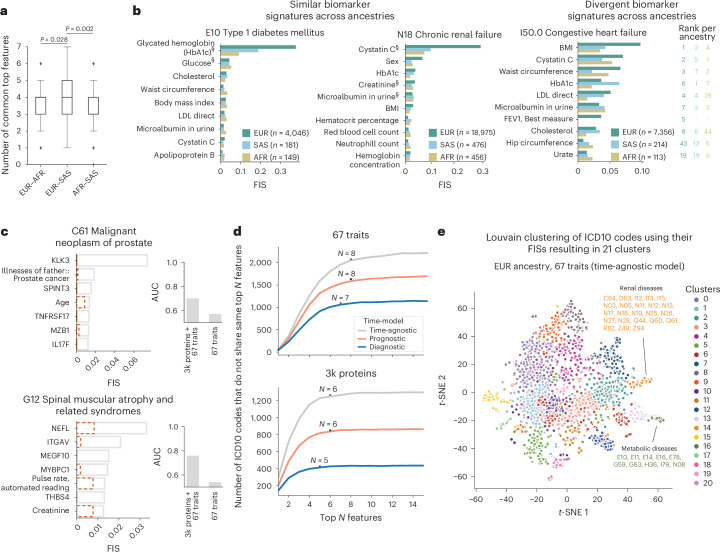
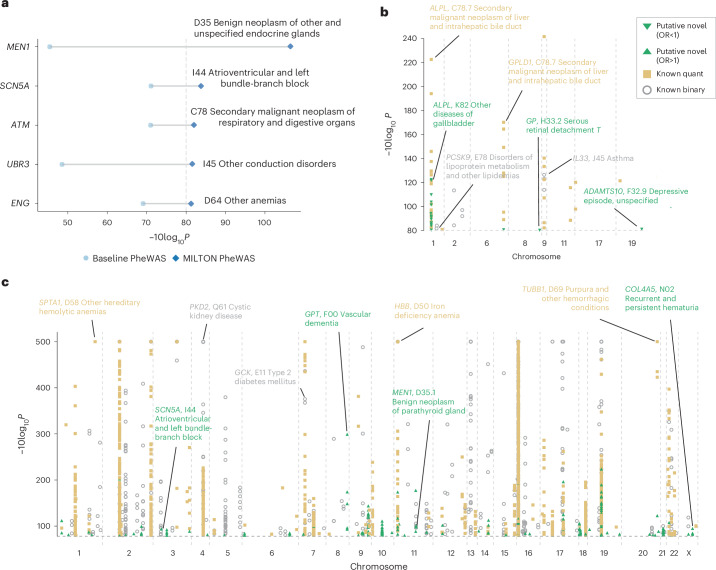
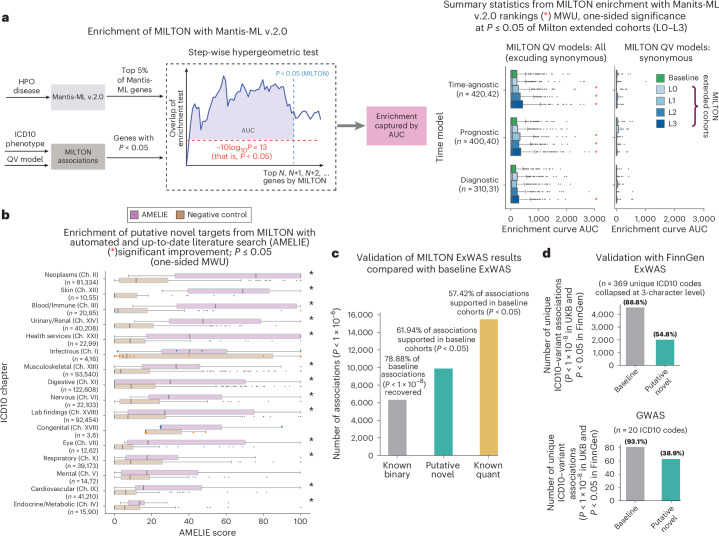
The emergence of biobank-level datasets offers new opportunities to discover novel biomarkers and develop predictive algorithms for human disease. Here, we present an ensemble machine-learning framework (machine learning with phenotype associations, MILTON) utilizing a range of biomarkers to predict 3,213 diseases in the UK Biobank. Leveraging the UK Biobank's longitudinal health record data, MILTON predicts incident disease cases undiagnosed at time of recruitment, largely outperforming available polygenic risk scores. We further demonstrate the utility of MILTON in augmenting genetic association analyses in a phenome-wide association study of 484,230 genome-sequenced samples, along with 46,327 samples with matched plasma proteomics data. This resulted in improved signals for 88 known (P < 1 × 10-8) gene-disease relationships alongside 182 gene-disease relationships that did not achieve genome-wide significance in the nonaugmented baseline cohorts. We validated these discoveries in the FinnGen biobank alongside two orthogonal machine-learning methods built for gene-disease prioritization. All extracted gene-disease associations and incident disease predictive biomarkers are publicly available ( http://milton.public.cgr.astrazeneca.com ).
© 2024. The Author(s).
Conflict of interest statement
M.G., M.K., D.M., L.M., O.S.B., F.H., E.W., K.R.S., M.A.F., J.M., A.O’N., E.A.A., A.R.H., Q.W., R.S.D., S.P. and D.V. are current employees and/or stockholders of AstraZeneca. E.A.A. is a founder of Personalis, Inc., DeepCell, Inc. and Svexa Inc.; a founding advisor of Nuevocor; a nonexecutive director at AstraZeneca; and an advisor to SequenceBio, Novartis, Medical Excellence Capital, Foresite Capital and Third Rock Ventures.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
